


Ehsan Larik, Muhammad Arif *, Ambreen Chaudhry, Muhammad Abdullah, Zubair Ahmed.
Field Epidemiology and Laboratory Training Program, Pakistan
*Corresponding Author: Muhammad Arif, Epidemiologist, FE(L)TP | National Institute of Health, Park Road, Islamabad 44000, Pakistan
Abstract
Xeroderma Pigmentosum is a rare genetic disorder inherited as an autosomal recessive pattern. Patients having the disease are intensely sensitive to ultraviolet (UV) rays. Exposure to sunlight leads to defective DNA repair and patients often suffer lesions on exposed body parts like the face, arms, and neck. Presentation of the disease ranges from photosensitivity, dry skin, freckles, premature skin aging, and a substantially high incidence of malignant lesions. There is no cure for the XP and the prevention can delay skin lesions. In this study, we described five cases of XP, two of which had developed malignancies, from one tribe in Baluchistan province. Cases were first notified from a tribal village through social media. Cases were clinically diagnosed and confirmed on histopathology for the development of complications like Basal cell and Squamous cell carcinomas. Outreach health education plan was implemented upon our recommendations along with provision of medication and rehabilitation services as well as counseling to avoid consanguineous marriages.
Keywords: Baluchistan; Case report; DNA repair; Kachi; Skin disease; Xeroderma Pigmentosum
Introduction
Background
Xeroderma Pigmentosum (XP) is a rare autosomal recessive disorder that is caused by a heterogeneous group of DNA resulting in a reduced ability of DNA repair [1]. The body areas exposed to the sunlight are poorly repaired by the XP cells. Patients with the disease are at a 2000 times higher risk of developing basal cell carcinoma and melanomas [2]. These UV-induced damages usually first appear as early as 2 months of age [3]. Depending on the affected gene, there are seven types of Xeroderma disease (XD) with different prevalence and severity and among them, XPA is the most common one. XP has shown equal incidence among males and females and across all races. The incidence ranges from ½.5M in the USA, and ½.3 M live births in Western Europe. A high incidence has been reported from Japan (1/20,000), North Africa, and Middle East [4].
Consanguineous marriages are responsible for the appearance of disease [5]. The risk of developing nonmelanoma skin cancer and melanoma skin cancers are as high as 10, 000 and 20,000 respectively underage of twenty [5]. Melanoma, Basal cell carcinoma, and squamous cell carcinoma are the most prevalent malignancies among XP patients [6]. XP is an extremely disfiguring disease that needs the expert dermatological intervention, early diagnosis and regular follow-up as well as rehabilitation facility for critical symptoms managements [7].
Five cases of rare skin disease were reported by social media from tribe X in village LundiKhosa, district Kacchiin Baluchistan. Later the disease was diagnosed as XP. Upon investigation it was found that 18 cases (11females, 61 %) were reported from the same tribe between 2009-2011. The median age was 8 years (range 1-31 years) with a 100 % fatality rate. In this study, we have described the five cases from the same tribe X of Baluchistan. Informed consent was taken from the parents/guardians before taking pictures, interview the children and their caretakers. A local language translator was hired for the purpose.
Case Presentations
Five cases of XerodermaPigmentosum were presented at the disease surveillance and response unit. All were female children. The median age of patients was six years (range: 5-12yrs.). Two cases (40 %) developed squamous cell and basal cell carcinomas confirmed by their histopathology reports. The parents of 4 (80 %) cases had consanguineous marriages. Ophthalmic involvement was found intwo (40 %) cases. The median age of developing signs and symptoms was 8 months. The incidence of XP in this tribe was 5.5/10,000 people and prevalence were 40/10,000 in the village. All the tribe member families had a low socioeconomic status with no girl has ever gone to school.
Patient 1 was six years old female child (figure 1) who developed freckles and papules on her face at the age of 8 months. The lesions spread over her whole body in the next few months. This was associated with photophobia and oral cavity involvement for the last 2 months on clinical examination. She had multiple pigmented papules and plaques on her face, dense lentigines involving the whole body. She had actinic cheilitis and a pink nodule of 1cm diameter on the tip of her tongue. Both eyes had cicatricial changes, ectropion, loss of lashes, and conjunctional congestion with retinal haziness. Hair was normal, though trimmed short by parents. Teeth were pigmented, associated with poor hygiene leading to dense plaque involving all teeth. She had shown no neurological symptoms. Squamous cell carcinoma was confirmed by her previous medical record.
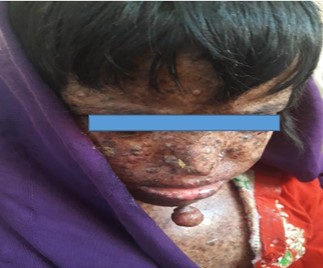
Figure 1: Six-year female diagnosed with Xeroderma Pigmentosum at the age of 8 months in Baluchsiatn-2019
Patient 2 was a five-year-old female (figure 2) developed first skin lesions at the age of 11 months. She showed stunted growth. Lentigines were found predominantly on photo-exposed parts with photophobia and conjunctional congestion.
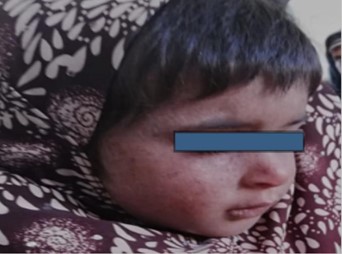
Figure 2: Five-year-old female diagnosed with XP at the age 11 months in Baluchistan-2019
Patient 3 was a twelve-year-old female (figure 3), malnourished, and was small for her age. Her father noticed skin lesions at six months of age. She underwent surgery for the removal of the nodule from the nose. The report of histopathology showed Basal Cell Carcinoma and squamous cell Carcinoma. She had dense lentigines on photo-exposed parts. She had exophthalmos of both eyes and complete loss of vision in the right eye from which a nodule was removed. Eyelashes were also absent.
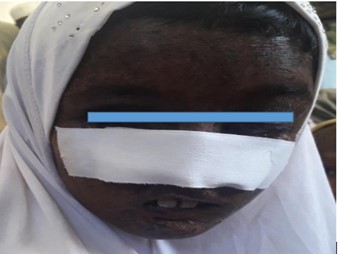
Figure 3: Twelve-year-old female diagnosed with XP at the age of six months in Baluchistan -2019
Patient 4 was a six-year-old female (figure 4) child who had multiple lentigines on exposed parts of body. She was having photophobia but no other eye signs and symptoms. Hair, nail, teeth, and neurological status were found normal. Her parents were from different tribes.
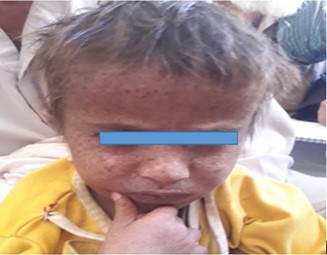
Figure 4: Six-year-old female diagnosed with XP whose parents are from two different tribes in Baluchistan-2019
Patient 5 was an 11-year-old female (figure 5) child whose parents noted skin changes at five months of age. She had multiple & dense lentigines on photo-exposed parts, thickening of facial skin multiple pigmented papules on the face, and a 1.5cm diameter nodule hanging from the lower jaw. Lips showed actinic changes and dyspigmentation, but oral mucosa and tongue were normal. Loss of eyes lashes, conjunctional congestion, mild retinal haziness was noted. Teeth, hair, nails were normal & no neurological deficit was noted.
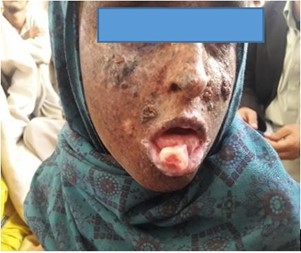
Figure 5: An 11-year-old female diagnosed with XP diagnosed at the age of 5 months in Baluchistan-2019.
Discussion
Tribal lifestyle, low literacy, and limited access to health care facilities along with prevailing myths about diseases are the major causes of complications and mortalities in the Baluchistan province of Pakistan [8]. Clinical features were found similar to 16cases reported 20 years back and were diagnosed as XerodermaPigmentosum according to narratives and the cases reported by other researchers where XPC gene expression was determined in impaired DNA repair mechanism upon sun exposure [9]. Though the history of consanguinity was present in all tribes of this region, only tribe X has shown the incidence of a cluster of XP. Severe ophthalmic manifestation affected all patients at an early age and the illness is largely a consequence of consanguineous marriages between close relatives over a number of generations. Consanguinity is prevalent in Pakistan and the rate varies among the urban and rural populations from 31% to 79% respectively. The tribal culture holds an even higher rate of interfamily marriage [10]. The high incidence of many otherwise rare disorders is solely due to consanguineous marriage like mental disorders and XP [11]. Contrary to our results, a study conducted in Zimbabwe showed no consanguinity at all. Rendering that cousin marriage is not an essential prerequisite for disease occurrence as is seen in one of these five cases [12].
The minimum age of expression in these cases was five months which much lower than that documented in similar cases in Turkey which were 1.4 years [13]. A study conducted in DI Khan Pakistan has shown the appearance of the first symptom as early as two months of age which suggests the varied expression of the disease based on a novel protein identification among these cases [4]. The mechanism of nucleotide excision (NER) repair deficit has been documented in two neurological diseases Xeroderma Pigmentosum (XP) and Cockayne syndrome (CS) however, none of these cases has a history of neurological problems [14]. Very early development of basal cell and squamous cell carcinoma was reported in two of the cases of age as low as 6 years. The risk of nonmelanoma skin cancer and melanoma inpatients with XP are more than 10,000 and 200 respectively and are shown to occur in patients younger than 20 years of age and are the most common cause of death [5].
Conclusion
Although there is no cure for XP early diagnosis, stringent sun protection, and regular follow-up can improve quality of life and life expectancy among cases with no neurological defects. Tribes were taught about covering windows with UV-resistant clothes and covering the whole body with white clothes.
Tribal lifestyle, prevailing misbelieves, and limited resources restricted the cases from seeking immediate medical care and led to high mortality previously. Genetic health education and psychological counseling play an important part in improving outcomes in remote areas. The authors suggest further studies on XP to improve prevention, early diagnoses, and rehabilitation.
Acknowledgment: We acknowledge the efforts of the health department, Government of Baluchistan for their support in the field investigation
Conflict of interest: The authors declared no conflict of interest
Funding for the study: No specific funding was awarded for the study
References
- Kraemer KH, Lee MM, Scotto J (1984) DNA repair protects against cutaneous and internal neoplasia: evidence from xeroderma pigmentosum. Carcinogenesis. 5(4): 511–4.
- Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, et al. (2011) Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 48(3): 168–76.
- Kocabalkan O, Ozgür F, Erk Y, Gürsu KG, Güngen Y (1997) Malignant melanoma in xeroderma pigmentosum patients: report of five cases. Eur J Surg Oncol. 23(1): 43–7.
- Ali MZ, Blatterer J, Khan MA, Schaflinger E, Petek E, et al. (2020) Identification of a novel protein truncating mutation p.Asp98* in XPC associated with xeroderma pigmentosum in a consanguineous Pakistani family. Mol Genet Genomic Med. 8(2): e1060.
- Gül Ü, Kılıç A, Gönül M, Çakmak SK, Soylu S (2007) Xeroderma pigmentosum: a Turkish case series. Int J Dermatol. 46(11): 1125–8.
- Tofuku Y, Nobeyama Y, Kamide R, Moriwaki S, Nakagawa H (2015) Xeroderma pigmentosum complementation group F: Report of a case and review of Japanese patients. J Dermatol. 42(9): 897–9.
- Dw E, Kd J, Joshi KD (2006) Xeroderma pigmentosa-A disfiguring disease. Kathmandu University Medical Journal. 4(1): 78-81.
- Bhutto AM, Shaikh A, Nonaka S (2005) Incidence of xeroderma pigmentosum in Larkana, Pakistan: A 7-year study. Br J Dermatol. 152(3): 545–51.
- Ijaz A, Basit S, Gul A, Batool L, Hussain A, et al. (2019) XPC gene mutations in families with xeroderma pigmentosum from Pakistan; prevalent founder effect. Congenit Anom (Kyoto). 59(1): 18–21.
- Wahab A, Ahmad M (1996) Biosocial perspective of consanguineous marriages in rural and urban Swat, Pakistan. J Biosoc Sci. 28(3): 305–13.
- Bittles AH (2001) Consanguinity and its relevance to clinical genetics. Clinical Genetics. 60(2): 89-98.
- Chidzonga MM, Mahomva L, Makunike-Mutasa R, Masanganise R (2009) Xeroderma Pigmentosum: A Retrospective Case Series in Zimbabwe. J Oral Maxillofac Surg. 67(1): 22–31.
- Akdeniz N, Güneş Bilgili S, Çalka Ö, Karadağ AS (2012) Xeroderma pigmentosum in eastern Turkey: a review of 15 cases. Turkish Journal of Medical Sciences. 42(4): 719-723
- Nouspikel T (2008) Nucleotide excision repair and neurological diseases. DNA Repair (Amst). 7(7): 1155–67.