


Víctor Raúl Luraschi Centurión1, Montserrat Almada Ruiz Diaz2, Roun Kim3*, Blas Antonio Romero Dure4
1Medical Director / Chief of General Surgery, Service ORCID: 0000-0002-2945-0852.
2Emergency Coordinator, General Surgery Service ORCID: 0000-0003-2125-239X.
3Specialist in General Surgery ORCID: 0009-0004-3502-3912.
4Pathologist ORCID: 0000-0003-3978-7042.
*Corresponding Author: Roun Kim, Consultant Neurosurgeon, Specialist in General Surgery ORCID: 0009-0004-3502-3912
Abstract
Castleman disease is a lymphoproliferative disorder, also known as angiofollicular lymph node hyperplasia. It is a rare condition with a wide range of clinical manifestations. We present the case of a 33-year-old male patient referred to our general surgery department due to a one- month history of ulcerated lesions on the distal extremities of both upper and lower limbs, as well as on the oral and genital mucosa, which were painful and progressively enlarging. These symptoms were accompanied by asthenia, weight loss, ocular pruritus, and conjunctival hyperemia. A computed tomography (CT) scan revealed a solid, hyper vascularized, well-defined neo proliferative mass with lobulated borders located in the left iliac fossa, measuring 8.4 × 7.4 × 5 cm (estimated volume of 162 cc), in close proximity to the external iliac vein. The patient underwent surgical resection of the tumor. Histopathological analysis confirmed the diagnosis of Castleman disease.
Keywords: Pemphigus, Paraneoplastic Pemphigus, Castleman Disease
Case Presentation
A 33-year-old male presented to another medical center with a one- month history of painful ulcerated lesions affecting the distal extremities (hands and feet), oral mucosa, and genital region. The symptoms were accompanied by asthenia, approximately 10 kg of weight loss, ocular pruritus, and conjunctival hyperemia. He was initially treated with prednisone (50 mg/day) and cyclosporine (200 mg/day), under the presumptive diagnosis of severe Stevens-Johnson syndrome.
Physical examination revealed oral ulcers (Figure 1) and genital involvement, including lesions on the prepuce. Ulcerative lesions were also observed on the distal aspects of the hands and feet (Figures 2A and 2B).
Initial laboratory workup showed: Hb 11.1 g/dL, Hct 33%, WBC 6,660/μL (71% neutrophils), platelets 280,000/μL, fibrinogen 476 mg/dL, Na/K/Cl 141/3.22/100 mEq/L, creatinine 0.72 mg/dL, AST/ALT 9/12 U/L.
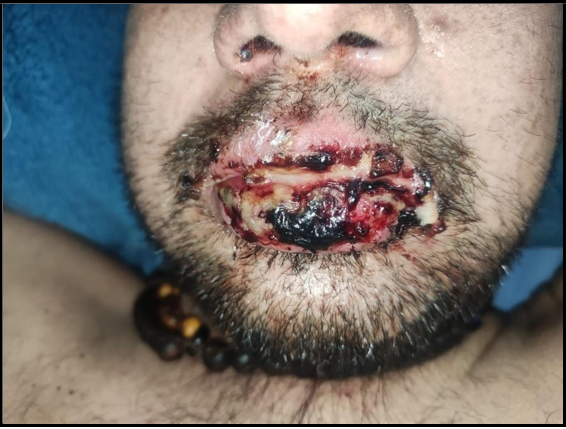
Figure 1: Severe oral Ulcer. Erosive cheilitis with serosanguinous scabs.
Given the suspicion of paraneoplastic pemphigus, a contrast- enhanced abdominal and pelvic CT scan was performed. The imaging revealed a solid, hyper vascularized, well- defined neo proliferative mass with lobulated borders, measuring 8.4 × 7.4 × 5 cm (162 cc) located in the left iliac fossa and in close relation to the external iliacvein (Figure 3).
The patient was then referred to our general surgery service for surgical biopsy of the mass, with a working diagnosis of paraneoplastic pemphigus of unknown origin.
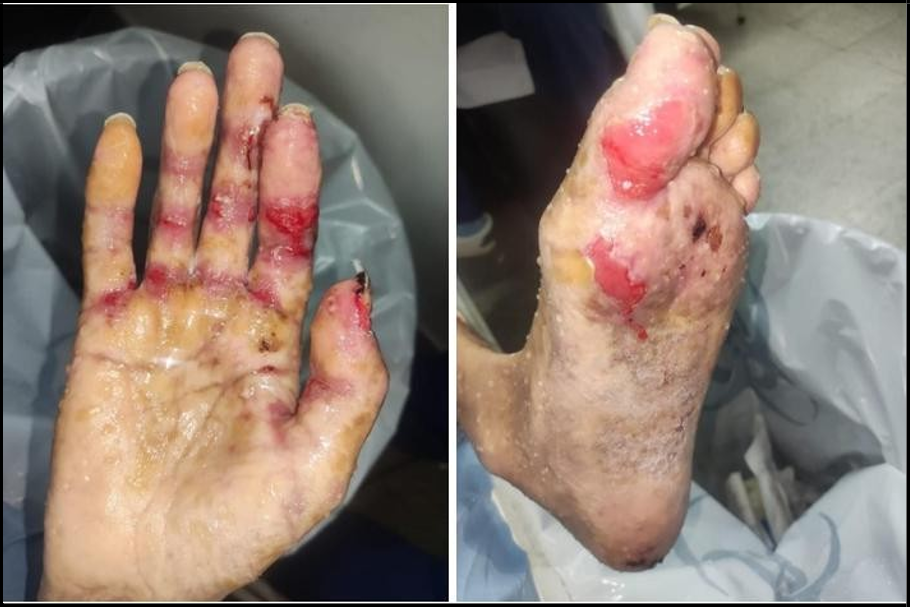
Figure 2A/B: Painful ulcers on the hands and the feet, with irregular borders and surrounded by an erythematous halo.
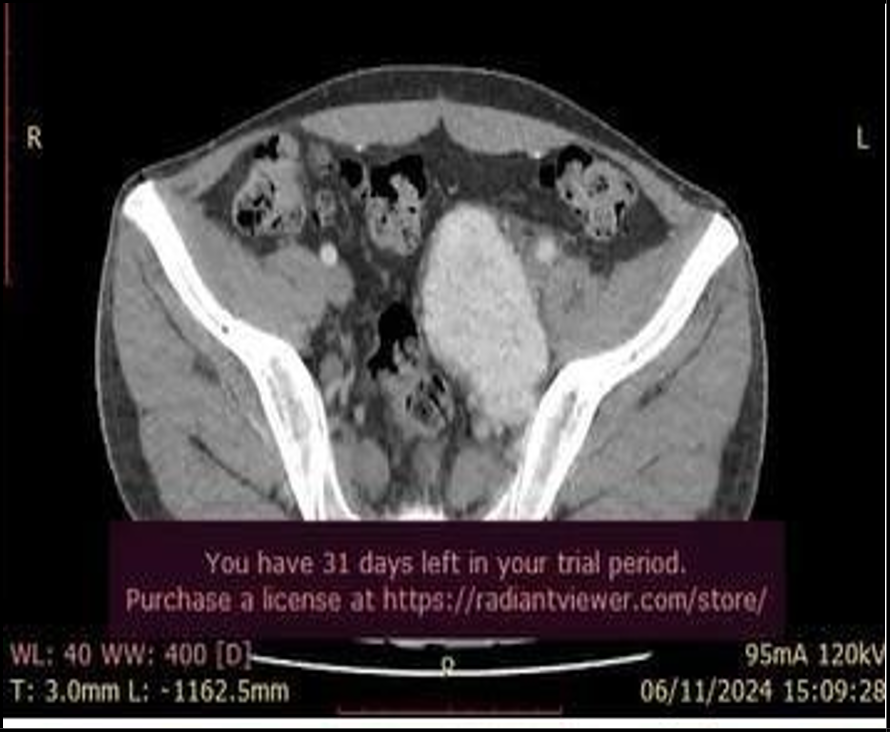
Figure 3: A solid, hypervascularized, well-defined neoproliferative mass with lobulated borders, measuring 8.4 × 7.4 × 5 cm (162 cc) located in the left iliac fossa and in close relation to the external iliac vein.
Intraoperative findings included a retroperitoneal mass in the left iliac fossa measuring approximately 9 cm in diameter, solid-elastic in consistency and with limited mobility, closely related to the iliacvessels. No enlarged iliac lymph nodes or free fluid in the abdominal cavity were noted. No additional lesions were identified (Figure 4).
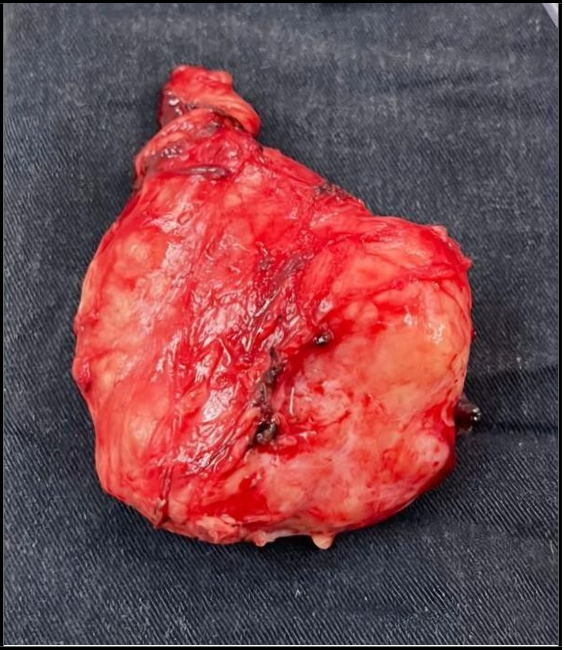
Figure 4: Mass in the left iliac fossa measuring approximately 9 cm in diameter, solid-elastic in consistency.
Histopathological examination confirmed a lesion compatible with Castleman disease, with differential diagnosis including other lymphoproliferative processes (Figures 5A, 5B, 5C). The patient was subsequently transferred back to his referring center for chemotherapy.
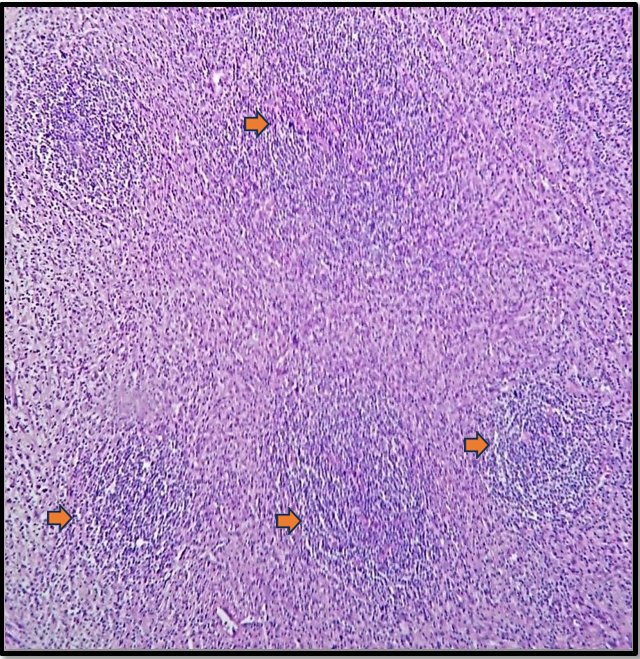
Figure 5A: Atretic germinal centers traversed by hyalinized and sclerotic penetrating vessels (arrows).
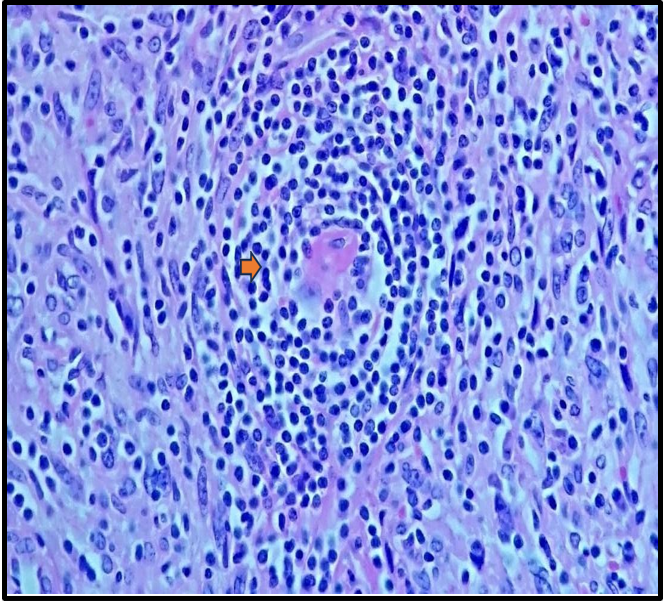
Figure 5B: The mantle zones are thickened, with lymphocytes arranged in concentric layers, creating an 'onion-skin' appearance (arrow).
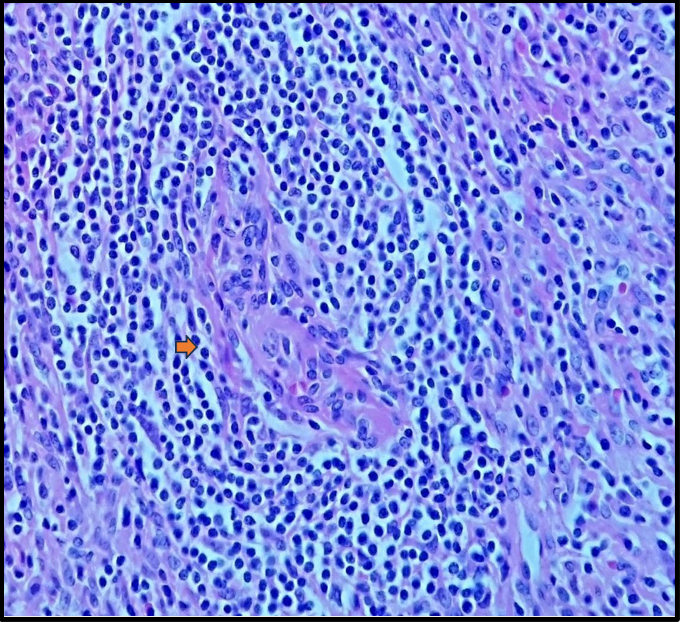
Figure5C: The mantle zones are thickened, with lymphocytes arranged in concentric layers, creating an 'onion-skin' appearance (arrow).
Discussion And Case Analysis
Castleman disease was first described in 1956 by Dr. Benjamin Castleman [1], an American pathologist. It is a benign, non-clonal lymphoproliferative disorder that is definitively diagnosed through histopathological examination. This rare and heterogeneous group of entities presents with a broad spectrum of clinical manifestations. [2] It has also been referred to as "angiofollicular lymph node hyperplasia" or "giant nodular hyperplasia." Castleman disease most commonly affects young adults, with a median age of 23 to 35 years for unicentric forms, and around 64 years for multicentric forms, with no clear gender predominance. [3, 4]
There are multiple etiologies, and the disease can affect any region of the body. The most frequently affected area is the thorax (70%), followed by the cervical region (15%), and the abdomen and pelvis (15%). [5]
Clinically, Castleman disease is classified into two subtypes:
1. Unicentric Castleman Disease (UCD), which is often paucisymptomatic. The most common histological variant is hyaline-vascular (80–90%).
2. Multicentric Castleman Disease (MCD), which often presents with systemic manifestations such as fever, weight loss, hypoproteinemia, generalized lymphadenopathy, and renal dysfunction. This subtype includes plasmacytic and mixed cell variants.
There are three major histological variants: hyaline-vascular (HV), plasmacytic, and HHV-8–associated. The original report by Dr. Castleman described the hyaline-vascular subtype, characterized by lymph nodes with atrophic or regressed germinal centers, often hyalinized, primarily composed of residual follicular dendritic cells and prominent mantle zones with small lymphocytes. These dendritic cells are arranged in concentric rings, creating the classic "onion- skin" appearance. The plasmacytic variant is the most common form of MCD (75%). [6] The coexistence of hyaline-vascular and plasmacytic features defines a subset of HHV-8–associated MCD. [7] In 1990, Anhalt et al. described a clinical entity known as Paraneoplastic Pemphigus (PNP), later defined by Nguyen et al. as a heterogeneous autoimmune syndrome affecting multiple organs. PNP is strongly associated with both benign and malignant tumors, most commonly lymphoid and hematologic malignancies such as B- cell lymphoma, chronic lymphocytic leukemia, Castleman disease, Waldenström's macroglobulinemia, and thymoma. [8]
We presented the case of a male patient with oral, genital, and extremity ulcerations in whom the diagnosis of PNP was established as an atypical presentation of multicentric Castleman disease.
Multicentric Castleman disease is aggressive and may be associated with conditions such as POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes), amyloidosis, Kaposi's sarcoma, renal failure, and HHV-8 or HIV infection. As previously mentioned, the most frequent locations are thoracic (70%), followed by cervical (15%), and abdominal/axillary regions. [9] In our case, the mass was located in the left iliac fossa, in close contact with the retroperitoneum—an uncommon location. The histologic subtype identified was hyaline-vascular (Figures 5A, 5B, 5C).
In our patient, the key clinical findings were mucocutaneous, consistent with a diagnosis of PNP. Notably, approximately 18% of PNP cases are associated with Castleman disease, although only a limited number of such cases have been reported. [10]
PNP is a severe autoimmune mucocutaneous disorder characterized by painful stomatitis, polymorphic skin eruptions, and underlying neoplasms. Histopathologically, it is defined by dyskeratotic epithelial cells, acantholysis, and typical immunofluorescence findings. [11] The prognosis of PNP is generally poor, with a mortality rate ranging from 75–90% and an average survival of less than one year. [12]
Camisa and Helm classified diagnostic criteria into major and minor signs:
1. Major criteria include polymorphous mucocutaneous eruptions, concurrent neoplasm, and the presence of serum autoantibodies with a specific immunoprecipitation pattern.
2. Minor criteria include histologic evidence of acantholysis, direct immunofluorescence showing intercellular and basement membrane staining, and indirect immunofluorescence using rat bladder epithelium.
Diagnosis requires three major criteria or two major plus two minor criteria. In our patient, two major criteria (polymorphous mucocutaneous eruption and concurrent neoplasm) and one minor criterion (histological acantholysis) supported the diagnosis of PNP. The most effective treatment for PNP is the resection or resolution of the underlying neoplasm, as has been demonstrated in cases associated with Castleman disease.
Mortality due to bronchiolitis obliterans, the most severe complication, appears to be reduced with early tumor resection and administration of immunoglobulin. Literature supports the use of high-dose systemic corticosteroids such as prednisone (1.0–1.5 mg/kg/day) and immunosuppressants like azathioprine or mycophenolate mofetil. [13] Rituximab has also been proposed to reduce B-cell–mediated antibody production responsible for skin lesions. [14] Our patient received high-dose corticosteroids without clinical improvement of the mucocutaneous lesions.
Castleman disease treatment is tailored to the underlying pathogenic mechanism. Surgical excision is the treatment of choice for UCD, whereas in MCD, cytoreductive therapy may be necessary. [15] Other treatments described in the literature include radiotherapy and immunomodulatory therapy (e.g., corticosteroids, interferon-α, all- trans retinoic acid, and thalidomide). [16, 17]
Following surgical tumor resection, our patient continued outpatient follow-up with Ophthalmology and ENT and was treated with azathioprine and corticosteroids.
Conclusion
Mucocutaneous lesions are common clinical findings in medical practice, often caused by infectious, autoimmune, or neoplastic processes. In cases unresponsive to antimicrobial or immunosuppressive therapies, it is essential to consider the possibility of a paraneoplastic syndrome. This case illustrates paraneoplastic pemphigus associated with Castleman disease. While the unicentric form of Castleman disease typically follows a benign course—as was observed in our patient—the multicentric form can present a more aggressive clinical trajectory. Histopathology plays a pivotal role in confirming the diagnosis and guiding appropriate treatment in such complex presentations.
Conflict Of Interest: The authors declare that there is no conflict of interest regarding the publication of this manuscript.
References
- CASTLEMAN B, IVERSON L, MENENDEZ VP (1956) Localized mediastinal lymph node hyperplasia resembling thymoma. Cancer. 9(4): 822-30.
- Bonekamp D, Horton KM, Hruban RH, Fishman EK (2011) Castleman disease: the great mimic. Radiographics. 31(6): 1793– 807.
- Maslovsky I, Uriev L, Lugassy G (2000) The heterogeneity of Castleman disease: report of five cases and review of the literature. Am J Med Sci. 320(4): 292-5.
- de Toledo Sancho JS, Sabaté JF, Irastorza CF, Layret XL, Fuentes NT, et al. (2005) Enfermedad de Castleman. An Pediatr (Barc). 63(1): 68-71.
- Keller AR, Hochholzer L, Castleman B (1972) Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 29(3): 670–83.
- Talat N, Belgaumkar AP, Schulte KM (2012) Surgery in Castleman’s disease: a systematic review of 404 published cases. Ann Surg. 255(4): 677–84.
- Szalat R, Munshi NC (2018) Diagnosis of Castleman Disease. Hematol Oncol Clin North Am. 32(1): 53–64.
- Tirado-Sánchez A, Bonifaz A (2017) Paraneoplastic pemphigus: a life-threatening autoimmune blistering disease. Actas Dermosifiliogr. 108(10): 902–10.
- Erkurt MA, Aydogdu I, Kuku I, Kaya E, Mizrak B, et al. (2009) A multicentric, hyaline vascular variant of Castleman's disease associated with B cell lymphoma: a case report. Cases J. 2: 8183.
- Bin Waqar SH, Khan AA, Mohiuddin O, Rehan A (2019) Paraneoplastic pemphigus with underlying Castleman’s disorder: a rare report with literature review. Cureus. 11(6): e5022.
- Zhu X, Zhang B (2007) Paraneoplastic pemphigus. J Dermatol. 34(8): 503–11.
- Camisa C, Helm TN (1993) Paraneoplastic pemphigus is an autoimmune disease induced by specific neoplasms. Arch Dermatol. 129(7): 883–6.
- Paolino G, Didona D, Magliulo G, Iannella G, Didona B, et al. (2017) Paraneoplastic pemphigus: insight into the autoimmune pathogenesis, clinical features and therapy. Int J Mol Sci. 18(12): 2532.
- Maruta CW, Miyamoto D, Aoki V, Carvalho RGR, Cunha BM, et al. (2019) Paraneoplastic pemphigus: a clinical, laboratorial, and therapeutic overview. An Bras Dermatol. 94(4): 388–98.
- Casper C (2005) The aetiology and management of Castleman disease at 50 years: translating pathophysiology to patient care. Br J Haematol. 129(1): 3–17.
- Bowne WB, Lewis JJ, Filippa DA, Niesvizky R, Brooks AD, et al. (1999) The management of unicentric and multicentric Castleman’s disease: a report of 16 cases and a review of the literature. Cancer. 85(3): 706–17.
- Soumerai JD, Sohani AR, Abramson JS (2014) Diagnosis and management of Castleman disease. Cancer Control. 21(4): 266– 78.