


Chamodika Lowe
Department of Biomedical Science, Faculty of Science and Engineering, University of Wolverhampton, Wolverhampton, U.K
*Corresponding Author: Chamodika Lowe, Department of Biomedical Science, Faculty of Science and Engineering, University of Wolverhampton, Wolverhampton, U.K
Abstract
DiGeorge Syndrome which is also known as chromosome 22q11.2 deletion syndrome is a primary immunodeficiency caused by the deletion of chromosome 22. Its main features include dysmorphia, hypoparathyroidism, hypocalcemia, hypoplasia or aplasia of the thymus, cardiac anomalies, renal anomalies, and behavioral/ psychiatric issues. This incurable syndrome could be treated for its complications to increase the quality of life. With the advancement of technology, DiGeorge syndrome can now be identified in childhood itself where FISH is the main diagnostic method used. A case report of a 5-year-old boy who visited the psychiatric hospital with attention deficit and socialization problems is presented here. The boy also had a medical history of mild recurrent numbness in his hands and showed mild facial dysmorphism on physical examination. On the evaluation of his case, Partial DiGeorge Syndrome was confirmed.
Keywords: Attention-deficit; Chromosome 22q11.2 deletion; DiGeorge syndrome; Dysmorphia; Hypocalcemia; Hypoparathyroidism; Microdeletion
Introduction
Background
DiGeorge Syndrome (DGS), as described in 1968 by Angelo DiGeorge is a primary immunodeficiency caused by abnormal development of 3rd and 4th pharyngeal pouches in the embryonic state [4]. This is due to microdeletions in sub-band 2 of band 1 in region 1 of the long arm of chromosome 22 where about 30-40 genes are deleted. Hence, DGS is also known as chromosome 22q11.2 deletion syndrome. This results in hypoplasia/ aplasia of thymus and parathyroid glands leading to immunodeficiency. Other main features of DGS include hypocalcemia, hypoparathyroidism, facial dysmorphism, cardiovascular anomalies, etc. [12].
DGS can be divided into two as partial DGS (pDGS) where the T-cell count will be low/ normal resulting in incomplete T-cell immunodeficiency and complete DGS (cDGS) where there will be no T-cells resulting in complete T-cell immunodeficiency [2]. Fluorescent in situ hybridization (FISH) is used to test DGS [3].
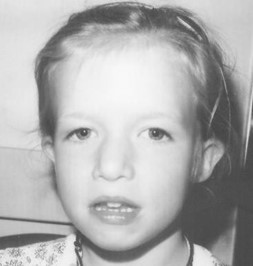
Figure1. A child with DGS [3]
Etiology and Epidemiology
DGS is the commonest of human genetic microdeletion syndromes. It can be seen in both males and females equally. Its occurrence is one in every 3000-6000 live births. Its diagnosis is done in early childhood, but with the unavailability of genetic testing before the 1990s there can be undiagnosed adults of age > 40yrs. Though there is no cure for DGS, early diagnosis and treatment of complications can increase the quality of life in patients [8].
The etiology of DGS is due to the haploinsufficiency of one or more genes on chromosome 22 in humans [4].
Though most of the DGS cases arise as de novo mutations, it can be inherited and shows an autosomal dominant inheritance pattern [12].
Case Presentation
A 5-year-old boy born to unrelated healthy parents was seen at a psychiatric hospital with attention deficit and socialization problems. Following psychiatric assessment, the patient was referred pediatric endocrinology department at the pediatric hospital for evaluation of possible hypothyroidism. The patient’s mother reported that he was inattentive and asocial. The boy had a medical history of mild recurrent numbness in his hands in the past 2 years. During the physical examination, mild facial dysmorphism was seen - prominent nose with a bulbous tip, smallmouth and eyes, ocular hypertelorism, and long face.
The laboratory investigations revealed a serum calcium level of 6.6 mg/dL (Normal 9-10.5 mg/dL), phosphorus 8 mg/dL (Normal: 2.4 to 4.1 mg/dL) parathormone level of 8 pg/mL (normal: 10-65), 25- hydroxyvitamin D level of 25 μg/L, and urine calcium/creatinine ratio of 0.01. His thyroid hormone levels were within the reference range. The thymus was not visualized at scintigraphic evaluation. Immunologic studies revealed normal levels (%) and absolute numbers (per mm3) of T cells/T-cell subsets, B cells, and NK cells, except for a slightly decreased CD3+ (CD16+56+)-cell level (48-58 %; normal range for children: 58-82 %). Ultrasonography of the left kidney demonstrated a cortical cyst. Fluorescence in situ hybridization (FISH) analysis, showed the chromosome 22q11.2 deletions. DiGeorge syndrome was diagnosed.
Case Details and Analysis
Clinical Findings and Analysis
According to the case study, the patient is a 5-year-old boy born to unrelated healthy parents. He has been following psychiatric assessment for attention deficit and socialization problems. It was also reported that he was inattentive and asocial. During the physical examination, it has been able to find mild facial dysmorphism – prominent nose with a bulbous tip, smallmouth and eyes, ocular hypertelorism, and long face. All these symptoms illustrate the characteristic features of DGS.
Deletion of chromosome 22q11.2 leads to the absence of the T-box1 (TBX1) gene which is involved in the development of 3rd and 4th pharyngeal pouches in the fetus which later gives rise to different parts of the head and neck, and hence, results in abnormalities in those parts and cause facial dysmorphism. TBX1 also causes the development of brain mesoderm, hence its absence may lead to neuropsychiatric issues. Small vermis and reduction in the gray matter will have resulted from the absence of TBX1 and causes socialization problems as it’s controlled by the posterior vermis and psychiatric issues, respectively [11].
Since the patient’s parents are unrelated and healthy, it confirms that the case is due to a de novo mutation and hasn’t been inherited.
The boy has a 2yrs medical history of mild recurrent numbness in his hands which can be due to hypocalcemia which is another feature of DGS [1].
Laboratory Findings and Analysis
Table 1: Interpretation of laboratory findings
|
Laboratory Investigation |
Result |
Normal Level |
Interpretation |
|
Serum Calcium |
6.6 mg/dL |
9 – 10.5 mg/dL |
PTH increases serum calcium level, but as PTH is low due to hypoparathyroidism, serum calcium is low. This is a characteristic feature of DGS. |
|
Phosphorus |
8 mg/dL |
2.4 – 4.1 mg/dL |
Due to the low level of PTH, reabsorption of phosphorus from kidneys increases increasing the serum phosphorus level. |
|
Parathormone (PTH) |
8 pg/mL |
10 – 65 pg/mL |
PTH is secreted by the parathyroid glands. The hypoplasia of parathyroid glands resulted from 22q11.2 deletion causes low levels of PTH. |
| 25-hydroxyvitamin D | 25 μg/L |
>20 μg/L [10] |
2-hydroxyvitamin D helps in the absorption of Calcium. Test confirming the normal level of 25- hydroxyvitamin D shows that a low level of serum Calcium isn’t due to an effect of 25- hydroxyvitamin D. |
| Urine Calcium/ Creatinine ratio | 0.01 |
<0.14 [10] |
The urine Calcium/ Creatinine ratio tends to increase in children with deficient vitamin D [10].Since this patient has a normal urine calcium/ creatinine ratio it tells that the patient has sufficient vitamin D. |
| CD3+ (CD16+56+) | 48-58% | 58-82% |
CD3+ (CD16+56+) value is slightly low which can lead to immunodeficiency in the patient but there is no significant evidence shown in the case for immunodeficiency. |
| Thyroid Hormones | Normal |
Sometimes thyroid hormones can be decreased due to low PTH levels and cause hypothyroidism. But since this patient has normal levels of thyroid hormones, he doesn’t have hypothyroidism. |
Laboratory investigations have revealed a low level of parathormone (PTH), low serum calcium, and a high level of phosphorus. This is a characteristic of DGS where PTH decreases due to hypoparathyroidism and causes Serum calcium to decrease and phosphorus to increase. Laboratory tests have also confirmed normal levels of 25-hydroxyvitamin D, Urine calcium/ creatinine ratio, and thyroid hormones as explained in Table-1.
Immunologic studies have revealed normal levels (%) and absolute numbers (per mm3) of T cells/T-cell subsets, B cells, and NK cells. A slight decrease in CD3+ (CD16+56+)-cell level has resulted which can lead to immunodeficiency. Also, the thymus hasn’t been visualized at scintigraphic evaluation.
But, since the T-cell count is normal it can be either that thymic tissues have been placed ectopically [2] or can be that T-cells are nonfunctional though the number is normal due to thymus hypoplasia/ aplasia.
Ultrasonography of the left kidney has demonstrated a cortical cyst which is again a feature of DGS.
FISH the analysis had shown the chromosome 22q11.2 deletions which confirm that the patient is having DiGeorge Syndrome.
Diagnosis
Patients don’t always have the characteristic dysmorphic facial features and so the standard diagnostic method of DGS is FISH [5]. But now with the advancement of technology, modern tests such as microarrays, single nucleotide polymorphism (SNP) arrays, and multiplex Polymerase Chain Reaction (PCR) methods are increasingly replacing FISH [8].
FISH is used to identify the submicroscopic heterozygous deletions of chromosome 22q11.2 using a probe specific for chromosome 22 and with a second probe that will hybridize the region commonly deleted. DGS is identified if this 2nd probe is absent. Disadvantages of FISH are that it is labor-intensive, needs specific equipment and training, expensive, turnover time is about 3-14days and mainly it cannot detect atypical deletions [11].

Figure2. FISH analysis with the Vysis N25 probe specific to Di-George region, arrow shows a deletion of chromosome 22q11.2 [7]
PCR is an alternative to FISH is more cost-effective and gives rapid results due to multiplexing [11].
SNP arrays are twice expensive and take a turnover time as same as FISH, but they can be used to identify atypical deletions too, and defects of other chromosomes can also be detected on the same assay [11].
Multiplex ligation-dependent probe amplification with high-density (MLPA–HD) is used to identify atypical deletions in 22q11 [4].
Prenatal ultrasound can be used to diagnose an affected fetus. Screening can be done through fetal echocardiography, chronic villi sampling, and amniocentesis [11].
Treatments and Prognosis
The mortality rate in DGS is unknown and though the rate of childhood deaths is rare, most of it has been due to cardiac diseases [11].
Therefore, early diagnosis of disease complications and management will lead to a better prognosis. Management of DGS is dependent on the patient’s age and phenotype, and treatment is personalized according to the complication severity.

Figure3. Hematoxylin and Eosin-stained biopsy of transplanted thymus viewed under low power. Thymic tissue is looking normal and is surrounded by striated muscle. A good corticomedullary distinction is observed. [2]
A common complication of DGS is hypocalcemia caused due to hypoparathyroidism which can be treated by treating for hypocalcemia by giving oral calcium supplements together with active vitamin D, while Teriparatide is a novel treatment. cDGS patients with complete immunodeficiency will have to be treated by performing a thymus tissue transplantation and T-cell transplantation while incomplete immunodeficiency can be treated with live vaccines like preventive measures for infections [6].
Patients with psychiatric disorders can be treated by psychotherapy together with medications [4].
Treatments for a few other complications are explained in Table-2.
Table 2: Treatments for DGS complication
|
Disease Complications |
Treatments |
|
Hypoparathyroidism |
Hypocalcemia and hyperphosphatemia arise due to hypoparathyroidism. Therefore, hypoparathyroidism can be treated by treating hypocalcemia. Treatment is giving supplements of calcium together with oral calcitriol which is active vitamin D. But should make sure not to do overtreatment because if so will lead to several other problems such as renal failure, hypercalciuria, nephrolithiasis, and nephrocalcinosis. hrPTH 1-34 (human recombinant parathyroid hormone) which is also called Teriparatide is being newly used to treat chronic hypocalcemia in children [6]. |
|
Incomplete Immunodeficiency |
Primary immunization [12]. |
|
Complete Immunodeficiency |
Thymus tissue transplantation; Fully matched T-cell transplantation; Immunoglobulin replacement therapy; Antiviral and antifungal treatments. Should avoid live-viral vaccines in severe immunodeficiency [6]. |
|
Cardiac anomalies |
Many cardiac anomalies have to be treated surgically [6]. |
| Recurrent Infections |
Otitis media is more common and can be treated by the placement of tympanostomy tubes like ventilation tubes. Should check for loosing of hearing using sound-field audiometry [3]. |
| Behavioral and Neuropsychiatric issues |
Psychotherapy. Anxiety and depression-like symptoms can be treated with medication [4]. |
|
Feeding difficulties in children |
Anti-reflux medications; Nasogastric tubes or gastrostomy feeding tubes [6]. |
| Palatal Defects | Surgery [6]. |
| Speech Difficulties | Speech [6]. |
Biochemical Aspects
Genetics
Most of the DGS cases have been reported due to de novo mutations while a lesser percentage is seen where an affected parent has passed the condition to the next generation showing an autosomal dominant pattern of inheritance [12].
Pathophysiology
Pathophysiology of DGS is by microdeletions on chromosome 22q11.2. This results in the deletion of 1.5-3 mb (million base pairs) including more than 30 genes that will get hemizygously deleted, and those deletions can be detected by molecular cytogenetic techniques such as FISH [8].
The cause for these deletions is by low copy number repeats (LCRs) and this region is comprised of four discrete LCRs blocks named LCR A-D. Asynchronous replication facilitated unequal meiotic exchange gives rise to these deletions [11].

Figure 4. The genes present within the commonly deleted region of chromosome 22 [11].
TBX1 and COMT (Catechol-O-methyl transferase) genes are two prominent genes deleted in 22q11.2 that causes the characteristic features of DGS such as hypocalcemia and neuropsychiatric disorders [6].
Non-Genetics Causes of DGS
Fetus getting exposed to retinoic acid will show phenotypes of DGS, as the expression of TBX1 in avian embryos, are affected by retinoic acid. Also, maternal diabetes and fetal alcohol syndrome will lead to DGS phenotypes [2].
Signs and Symptoms
Characteristic features of DGS include Facial dysmorphism – fish- mouth appearance, micrognathia, hypertelorism; hypocalcemia; hypoparathyroidism; cardiac abnormalities, hyperactivity/ attention deficit, renal defects, palatal defects and etc. [5].
Table3: Signs and Symptoms of DGS complication [11]
|
Feature |
Signs and Symptoms |
|
Dysmorphia |
Hooded eyelid; Bulbous nasal tip; Nasal dimple; Micrognathia; Microtia; Posteriorly rotated ears |
|
Cardiac anomaly |
Tetralogy of Fallot; Ventriculoseptal defect; Interrupted aortic arch; Truncus arteriosus; Vascular ring. |
| Immune deficiency |
T-cell lymphopenia; Delayed IgG production; Thymic aplasia with absent T cells. |
| Palatal defects |
Velopharyngeal insufficiency; Submucous cleft palate; Overt cleft palate; Cleft lip and palate |
| Hypoparathyroidism | Hypocalcemia, Hyperphosphatemia |
| Hypocalcemia | Muscle cramps; Numbness; Tetany; Seizures; Hypotension. [1] |
| Renal anomaly |
Structural renal anomaly; Renal agenesis; Dysplastic kidneys; Hypoplastic kidneys; Hydronephrosis |
| Hearing loss | Conductive hearing loss; Sensorineural hearing loss |
| Behavioral/psychiatric issue |
Generalized anxiety; Phobias; Attention deficit hyperactivity disorder; Autism |
| Gastrointestinal |
Intestinal malrotation; Hirschsprung; Tracheoesophageal fistula; Esophageal atresia |
| Endocrine | Hypothyroidism Growth hormone deficiency |
Biochemistry
TBX1 gene encodes for the development of embryonic 3rd and 4th pharyngeal pouches which will later give rise to different parts of the head and neck, thymus, parathyroid glands and great vessels of the heart [2].
TBX1 is also involved in brain mesoderm development. Due to the deletion of 22q11.2, TBX1 will be deleted causing abnormalities in the development of the pharyngeal pouches and results in hypoplasia or aplasia of parathyroid glands and thymus. Abnormal development in the pharyngeal pouches leads to defects in morphologies different organs that are derived from them and in parts of head and neck giving rise to characteristic features of DGS [11].
Hypoplasia of parathyroid glands causes it to produce a low level of parathyroid hormone/ parathormone (PTH) leading to hypoparathyroidism.
PTH is involved in regulating Serum calcium and phosphorous balance. Since PTH level is low, it causes the serum calcium level to decrease causing hypocalcemia and phosphorous level to increase causing hyperphosphatemia [9].
25-hydroxyvitamin D is converted into Calcitriol which is the active form of vitamin D, in the Kidney. Calcitriol assists the absorption of Calcium and if this calcitriol is deficient in children it will lead to an increase in urine calcium/ creatinine ratio [10].
Sometimes hypothyroidism can be caused in DGS due to loss of genes common to both thyroid gland and parathyroid glands [9].
Immunological Aspects
The thymus is derived from the 3rd pharyngeal pouch and TBX1 encodes transcription factors needed for the development of the thymus. Deletion of TBX1 due to 22q11.2 deletion causes hypoplasia or aplasia of the thymus. This will lead to primary immunodeficiency in DGS patients. Depending on the number of T-cells DGS can be either cDGS or pDGS. The aplastic thymus will lead to the absence of T-cells leading to complete immunodeficiency and will cause cDGS while the hypoplastic thymus can have T-cells numbers and functions from normal or near-normal to near-complete deficient leading to incomplete immunodeficiency causing pDGS. There can be conditions where T-cell numbers can be normal or near-normal in conditions where thymic tissues are placed ectopically [2].
Discussion
As discussed so far, DGS is caused by microdeletions of chromosome 22 located as 22q11.2 and can be identified using FISH analysis [12].
Similarly, the patient in the presented case study has been identified to be having DGS, with the use of FISH analysis.
Common phenotypic features of DGS include neuropsychiatric and behavioral disorders including hyperactivity/ attention deficit, socialization problems, hypocalcemia, renal anomalies, Facial dysmorphism, etc. [11].
This patient has also reported attention deficit, socialization problems, and facial dysmorphism including a prominent nose with a bulbous tip, smallmouth, and eyes, ocular hypertelorism, and long face, which are characteristic features of DGS resulted due to the deletion of the TBX1 gene.
Neuropsychiatric problems are resulted due to abnormal development of brain mesoderm due to deletion of TBX1 gene and also due to deletion of COMT gene. Socialization problems have resulted from the deletion of TBX1 leading to the development of a small vermis due to the reduction in gray matter, as psychiatric issues are maintained by the posterior vermus [11].His laboratory reports revealed low levels of serum calcium and a high level of phosphorus together with a low level of PTH which further confirms DGS.
One major feature of DGS is hypoparathyroidism which is revealed with patients having low PTH. Hypoparathyroidism causes hypocalcemia (low serum Calcium) and hyperphosphatemia (high phosphorus) and that can be clearly seen in the patient’s laboratory findings [9].
Therefore, he can be treated with calcium supplements together with active vitamin D.
The patient has a medical history of recurrent numbness in the hands and that is due to hypocalcemia [1].
The patient has a normal level of T-cells, but the thymus wasn’t visualized in the scintigraphic evaluation and hence, that can be due to an ectopic thymus tissue placement [2].
Though CD3+ (CD16+56+) cell level is slightly low which may lead to immunodeficiency, since B cells and NK cells are also normal and there is no significant evidence of immunodeficiency, the patient can be confirmed to be having pDGS.
Conclusion
According to the details given in the case study and clinical and laboratory findings, it’s able to conclude that the patient is having Partial DiGeorge Syndrome which has resulted due to a de novo mutation. His condition is not severe and can be treated for his disease complications such as with calcium and Calcitriol supplements for hypocalcemia, as discussed above.
References
- Al-taie N, Scheuter-Mlaker S, Schlesinger M, Abrahamian H (2014) Case report: DiGeorge syndrome presenting with hypoparathyrodism and Learning Difficulties in adulthood. British Journal of Medical Practitioners. 7(4): a730.
- Davies EG (2013) Immunodeficiency in DiGeorge Syndrome and Options for Treating Cases with Complete Athymia. Frontiers in Immunology. 4: 322.
- Dyce O, McDonald-McGinn D, Kirschner RE, Zackai E, Young K, et al. (2002) Otolaryngologic Manifestations of the 22q11.2 Deletion Syndrome. Arch Otolaryngol Head Neck Surg. 128(12): 1408-1412.
- Fomin ABF, Pastorino AC, Kim CAe, Pereira CA, Carneiro- Sampaio M, et al. (2010) DiGeorge Syndrome: a not so rare disease. Clinics. 65(9): 865-869.
- Hacıhamdioğlu B, Berberoğlu M, Şıklar Z, Doğu F, Bilir P, et al. (2011) Two Patients with Partial DiGeorge Syndrome Presenting with Attention Disorder and Learning Difficulties - Case Report. J Clin Res Pediatr Endocrinol. 3(2): 95-97.
- Hacıhamdioğlu B, Hacihamdioglu D, Delil K (2015) 22q11 deletion syndrome: current perspective. The Application of Clinical Genetics. 8: 123-132.
- Johnston PC, Donnelly DE, Morrison PJ, Hunter SJ (2008) DiGeorge syndrome presenting as late onset hypocalcaemia in adulthood. The Ulster Medical Journal. 77(3): 201-202.
- Kraus C, Vanicek T, Weidenauer A, Khanaqa T, Stamenkovic M, et al. (2018) DiGeorge syndrome: Relevance of psychiatric symptoms in undiagnosed adult patients. Wiener klinische Wochenschrift. 130(7-8): 283-287.
- Mantovani G, Elli FM, Corbetta S (2017) Hypothyroidism associated with parathyroid disorders. Best Practice & Research Clinical Endocrinology & Metabolism. 31(2): 161-173.
- Marwaha Rk, Garg Mk, Dang N, Mithal A, Narang A, et al. (2019) Reference range of random urinary calcium creatinine ratio in North Indian children and adolescents. Annals of Pediatric Endocrinology & Metabolism, 24(1):34-40.
- McDonald-McGinn DM, Sullivan KE (2011) Chromosome 22q11.2 Deletion Syndrome (DiGeorge Syndrome/Velocardiofacial Syndrome). Medicine. 90(1): 1-18.
- Nepesov S, Aygün FD, Küçüksezer U, Taşdemir E, Çokuğraş H, et al. (2019) Clinical and immunophenotypic characteristics of patients with chromosome 22q11.2 deletion syndrome: a single institution’s experience. Turk Pediatri Ars. 54(1): 28-34.