


Priscila Cardoso Braz Ascar1*, Giselle Machado Campos de Oliveira1, Fernanda Ghilardi Leão1, Luciano Silveira Onofre1, Jovelino Quintino de Souza Leão2, José Carnevale1
1Pediatric Urology Division, Hospital Infantil Darcy Vargas, São Paulo – São Paulo, Brazil.
2Head of Pediatric Urology Division, Hospital Infantil Darcy Vargas, São Paulo – São Paulo, Brazil.
*Corresponding Author: Priscila Cardoso Braz Ascar, Pediatric Urology Division, Hospital Infantil Darcy Vargas, São Paulo – São Paulo, Brazil.
Abstract
Introduction: Caudal Regression Syndrome (CRS) is an association of rectal, genitourinary, lumbosacral, and lower extremities anomalies with an extensive spectrum of presentations. As an early misdevelopment phenomenon, anatomical findings in this anomaly are vast, so individualized surgical treatment should be planned.
Methods: Between January 2010 and December 2021, medical charts of patients with anorectal malformation (ARM), lumbosacral or spinal cord anomalies, lower extremities, and genitourinary malformation were retrospectively reviewed. Demographic and clinical features were identified, and urological procedures performed in each patient, including urinary and bowel stomas, were described.
Cases presentation: Among 288 ARM patients, 19 (6,6%) were identified. Anorectal agenesis without fistula was the most frequent type of ARM (21,0%). Renal dysplasia and renal hypotrophy were found in 6 (31,5%).
Conclusions: CRS requires a multidisciplinary approach and early identification of patients with high intestinal obstruction and renal failure risk. The surgical team may have skills regarding complex organs and systems reconstruction.
Keywords: Caudal Regression Syndrome, anorectal malformation, dysraphism, neuropathic bladder dysfunction, case series.
Introduction
Caudal Regression Syndrome (CRS) is an association of varying anomalies of the rectum, urinary and genital tracts, lumbosacral spine, and lower extremities [1], which results in an extensive anatomical spectrum. Its definition is complex, sometimes making the distinction of patients with anorectal anomaly (ARM) associated with lumbosacral spine malformation difficult. Other terminologies are also used to define this syndrome, such as caudal dysgenesis syndrome, sacral agenesis, sacral dysgenesis or regression, sacrococcygeal dysgenesis, and caudal dysplasia sequence [2,3,4], but there is no agreement about the best terminology to be used in literature.
Low ARM and sirenomelia are extremes of an embryonic defect in the formation of the caudal region, the last being considered the most severe form of presentation of CRS by some authors [5]. Myelomeningocele, cloacal exstrophy, and Currarino syndrome are considered differential diagnoses of CRS. However, all these anomalies can be different manifestations of a single pathogenic process.
CRS affects 1 to 3 individuals out of 100,000 born-alive infants [4], and family history and maternal diabetes must always be considered [4,6]. From an embryonic perspective, CRS is thought to result from an abnormal gastrulation process before the fourth week of pregnancy and affects the mid-posterior axial mesoderm of the embryo [7] without a distinct, well-defined etiological agent. Phenotype changes present in each individual may be related to the insult and the moment when it occurs, besides other associated factors. Nowadays, it is thought that genetic and environmental factors contextualize this clinical condition and factors related to embryo vascularization in this development stage [8]. Mutations of VANGL1 genes in chromosome 1p13, CELSRI, and HLXB9 in chromosome 7q36 have been described [4]; however, as most cases are rare, it is thought that each represents an autosomal dominant condition as the result of spontaneous mutation [9].
The distinction of CRS is challenging, leading many times to mistaken definitions. The terminology caudal dysplasia sequence is the most proper one to determine this extensive spectrum of anomalies since there is no predictable pattern of severity in presentation.
Besides the high incidence of neuropathic bladder dysfunction, which occurs due to spinal malformation, renal dysplasia, renal anomalies of fusion and position, hydronephrosis, and vesicoureteral reflux (VUR) are frequent in CRS patients [10]. Urological assessment is vital to prevent recurrent urinary tract infections and renal impairment. The upper and lower urinary tract malformations are regularly associated with genital malformations involving gonads and internal and external genitalia. The main objective of this article is to report the experience of a Pediatric Urology Group and describe the genitourinary anomalies and surgical procedures used to treat CRS patients.
Methods
Study delineation
Accessible, informed consent was applied after the Research Ethics Committee submission and approval. A retrospective review of medical charts of all patients with ARM cared at Hospital Infantil Darcy Vargas was done between January 2010 and December 2021. Among them, patients with the association lumbosacral or spinal cord malformation, lower extremities deformities, and genitourinary tract anomalies reached the inclusion criteria for CRS. Associated syndromes such as Currarino syndrome, cloacal exstrophy, VACTERL Syndrome, and Prune Belly Syndrome were excluded.
Demographic and clinical features analyzed were gender, ARM, urinary tract, lumbosacral, spinal cord, genital and lower extremities anomalies, chronic kidney disease, and neuropathic bladder dysfunction. Moreover, urological procedures and types of urinary and bowel stomas were described in each patient.
Case Series
An overall of 288 patients were diagnosed with ARM. Among them, 19 (6,6%) were identified as having the inclusion criteria for CRS, 9 female (47,4%) and 10 males (52,6%). Concerning the type of ARM, 4 patients had anorectal agenesis without fistula (21,0%), 3 patients had a rectourethral fistula (15,8%), 3 were cloacas (15,8%), 2 had perineal fistula (10,5%), 2 rectovestibular fistula (10,5%), 2 urogenital sinus and anterior anus (10,5%), 1 rectovaginal fistula (5,2%) and 2 anterior anus (10,5%) (Table 1).
Table 1: Type of ARM, urinary tract, genitalia and lower extremities alterations.
|
Patient |
Type of ARM |
Urinary tract |
Genitalia |
Lower extremities |
|
1 |
Perineal fistula |
Renal hypoplasia |
Normal |
Clubfoot |
|
2 |
Perineal fistula |
Ectopia Horseshoe kidney |
Normal |
Fusion of popliteal fold Hypoplasia |
|
3 |
No fistula |
Fused pelvic kidney Megaureter |
Transposition Transverse testicular ectopia |
Bilateral clubfoot |
|
4 |
Rectourethral |
Horseshoe kidney |
Normal |
Genu valgum |
|
5 |
No fistula |
Pelvic kidney |
Transposition Hypospadia Testicular dystopia |
Hypoplasia |
|
6 |
Cloaca |
Right renal agenesis |
Cloaca |
Asymmetry |
|
7 |
Rectourethral |
Renal agenesis Hydronephrosis |
Normal |
Asymmetry, hypoplasia |
|
8 |
Rectovaginal |
Bilateral ureterohydronephrosis |
Normal |
Asymmetry Valgus and varus of the toes |
|
9 |
Urogenital sinus + anterior anus |
Renal hypoplasia Ectopic ureter |
Urogenital sinus |
Bilateral clubfoot |
|
10 |
Rectourethral |
Renal hypoplasia |
Transposition Bilateral testicular dystopia |
Asymmetry |
|
11 |
Undefined |
Right renal agenesis |
Transposition Testicular dystopia |
Asymmetry Anomalous implantation of the toes |
|
12 |
Cloaca |
Horseshoe kidney Duplicity VUR |
Cloaca Müllerian malformation |
Clubfoot Asymmetry |
|
13 |
No fistula |
Normal |
Transposition Testicular dystopia |
Bilateral clubfoot Ankle ankylosis |
|
14 |
No fistula |
Right renal agenesis |
Normal |
Bilateral clubfoot |
|
15 |
Cloaca |
VUR |
Cloaca |
Bilateral clubfoot Asymmetry |
|
16 |
Urogenital sinus + anterior anus |
Bilateral ureterohydronephrosis, VUR |
Urogenital sinus Müllerian malformation |
Right clubfoot |
|
17 |
Rectovestibular |
Left renal agenesis |
Asymmetry of labia majora |
Left clubfoot |
|
18 |
Undefined |
Right hydronephrosis |
Normal |
Asymmetry |
|
19 |
Rectovestibular |
Hydronephrosis |
Normal |
Hypoplasia of left lower extremity |
Urinary tract anomalies found in this cohort were renal dysplasia and hypotrophy in 6 (31,5%), 6 hydronephrosis (31,5%), 5 solitary kidney (26,3%), 3 horseshoe kidney (15,8%), 3 other renal anomalies of fusion and position (15,8%), 3 vesicoureteral reflux (VUR) (15,8%), 1 duplex collecting system and ectopic ureter (5,2%). Most patients presented with normal external genitalia (8 - 42,1%). Genital anomalies found were penoscrotal transposition in 5 boys (26,3%), undescended testis 5 (26,3%), one transverse testicular ectopia (5,2%), 3 genital hypoplasia (15,8%), 3 cloacas (15,8%), 2 urogenital sinus (10,5%), 2 Müllerian malformations (10,5%) and one hypospadias (5,2%). All patients presented neuropathic bladder dysfunction (19 -100%), and 12 (63,1%) patients developed some degree of chronic kidney disease.
Vertebral anomalies (Figure 1) were not always followed by spinal cord malformations (Table 2). Magnetic resonance images (MRI) were not performed in five patients. Surgical procedures for each patient varied from 1 to 5 interventions. These procedures were performed at our hospital and elsewhere.

Figure 1: Computed tomography of the pelvis with vertebral alteration - sacral agenesis
Table 2: Spinal cord and spine alterations
|
Patient |
Spine |
Spinal cord |
|
1 |
Deformity of the sacrum |
Sirenomelia |
|
2 |
Sacral agenesis |
Low-lying cord |
|
3 |
Fusion failure Deformity of the sacrum Hypoplasia of the sacrum |
Normal |
|
4 |
Sacral agenesis Transitional vertebrae L5 – S1 |
Sirenomelia Spinal cord with sudden disruption |
|
5 |
Sacral agenesis |
Myelomeningocele |
|
6 |
Sacral agenesis |
Lipomeningocele |
|
7 |
Sacral agenesis Scoliosis |
Normal |
|
8 |
Hypoplasia of the sacrum Coccyx agenesis Scoliosis |
Normal |
|
9 |
Sacral agenesis Hemivertebra in L1 |
Normal |
|
10 |
Hypoplasia of the sacrum Scimitar sign |
No evaluation |
|
11 |
Fusion failure Sacral agenesis |
No evaluation |
|
12 |
Deformity of the sacrum |
No evaluation |
|
13 |
Deformity of the sacrum |
Lipomeningocele |
|
14 |
Fusion failure |
Lipoma of the filum terminale |
|
15 |
Fusion failure Deformity of the sacrum |
Tethered cord with low-lying conus |
|
16 |
Deformity of the sacrum |
No evaluation |
|
17 |
Fusion failure |
No evaluation |
|
18 |
Deformity of the sacrum Coccyx agenesis |
Conus medullaris with sudden disruption |
|
19 |
Deformity of the sacrum Fusion failure |
Lipoma of the filum terminale |
All patients presented urinary bladder dysfunction, so they started clean intermittent catheterization (CIC) early in infancy. Among them, 12 underwent vesicostomy (63,1%), 7 of them (36,8%) had continent urinary stoma with Mitrofanoff conduits done afterward (Figure 2) and 5 (26,3%) maintained incontinent urinary stoma (4 vesicostomy and 1 nephrostomy). Three patients were submitted to bladder augmentation (15,7%), one of them as preparation for kidney transplantation. Concerning intestinal stoma, 6 patients (31.5%) still have colostomy. Eleven (57,8%) patients had already had colostomy closed, and 2 (10,5%) had Malone antegrade continence enema (MACE) stoma done (Figure 2).
Considering the correction of genital anomalies done so far, orchidopexy was the most frequent in 4 patients (21%), vaginoplasty in 2 patients (10,5%), penoscrotal transposition correction in 2 (10,5%), and urethroplasty in two (10,5%). Video laparoscopy for internal genitalia evaluation was done in 2 patients (10,5%), gonadal biopsy in 2 (10,5%), and Müllerian malformation resection was necessary in one (5,2%).
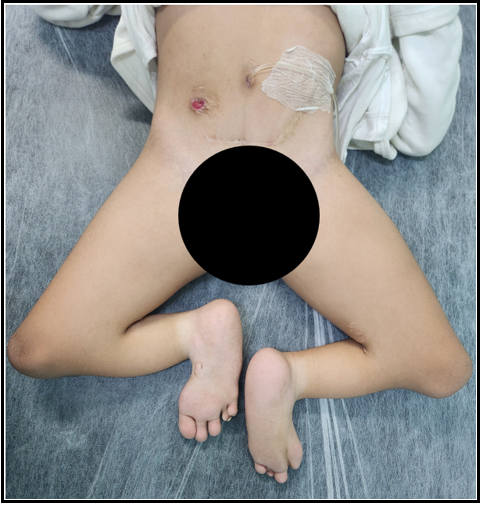
Figure 2: SRC patient with Mitrofanoff urinary stoma and Malone antegrade continence enema stoma
Discussion
In 1852, Geoffroy Saint-Hilaire and Hohl [3] described a series of malformations of multiple systems, and in 1910, Joshi Yadav associated this series of alterations with the absence of lumbosacral spine [11]. However, the term CRS was used for the first time in 1964 by Bernard Duhamel, who defined it as a spectrum of congenital malformations that included from asymptomatic agenesis of the coccyx in its most benign form up to sirenomelia as the most severe form [11,12]. Nevertheless, there is no agreement to define precisely what this syndrome consists of, leading to late diagnosis and treatment, consequently increasing the risk of renal injury, recurrent urinary tract infections, and incontinence [13].
Nerve fibers from S2 to S5 have autonomic, sensory, and motor functions and are also responsible for the anal and urinary sphincters. It is thought that the progression of neurologic injury frequently observed in children with CRS is due to inappropriate growth of sacral nerve roots that are in an unnatural position [13].
In patients with ARM, the association of sacral underdevelopment, tethered spinal cord, and urologic anomalies is well established [14], and this is also true for patients with CRS. Renshaw has set up a way to classify this syndrome according to the abnormal anatomy of the sacroiliac region [11]. In 1993, Pang revised this classification and applied it to 34 cases of lumbosacral agenesis [15]. However, a type that involves all CRS features and considers its functional prognosis is still necessary.
Patients with caudal region anomalies may have lumbosacral spine or spinal cord malformations, deformities in lower extremities, and genitourinary tract anomalies. This study aims to classify patients with these associated malformations as CRS complex, avoiding late diagnosis and treatment. These children will require a careful rehabilitation and functional maintenance approach from the neonatal period.
As the primary strategy for CRS treatment, most affected patients must be recognized after birth, and at this moment, ARM surgical treatment is prioritized because intestinal obstruction needs urgent resolution. Strict control of urinary output and kidney and collecting system ultrasound (US) investigation must be performed early to identify patients who can develop renal failure due to urinary anomalies, such as solitary kidney and obstructive hydronephrosis. In addition, this group of patients may require specialized investigation regarding gender definition based on external genitalia anomalies. Birth in referral centers facilitates a multidisciplinary approach when prenatal diagnosis is suspected. Genetic and imaging tests to identify lumbosacral and spinal cord anomalies are part of the therapeutic arsenal needed. Diagnostic laparoscopy should be performed to investigate gonadal and internal genitalia anatomy.
Bowel and urinary diversion, when indicated, should be considered early in surgical management to avoid potentially dangerous evolution. Further surgical interventions should be needed to offer urinary and fecal continence as these children grow. Some patients required continent urinary diversion and augmentation cystoplasty during childhood to achieve social continence. One patient who evolved to end-stage renal disease was submitted to Mitrofanoff conduit procedure instead of incontinent vesicostomy before kidney transplantation.
Chronic renal failure due to neuropathic bladder dysfunction and recurrent urinary tract infections may evolve into renal replacement therapy. Furthermore, multidisciplinary treatment of CRS patients, including urological, neurological, orthopedics, and anorectal reconstruction regarding the functional preservation of all systems involved, is essential for a better quality of life.
This study has its limitations, primarily due to its retrospective nature. Further studies, especially comparing patients with isolated ARM with those with CRS, would be relevant.
Conclusion
While the actual cause for CRS phenomenon development is widely discussed, a classification that involves all CRS anomalies and considers its functional prognosis is essential. This study selected patients with ARM, spinal cord or lumbosacral malformation, lower extremities deformities, and genitourinary anomalies as CRS. Severe practical consequences of untreated neuropathic bladder dysfunction may become chronic kidney disease. Urinary abnormalities such as hydronephrosis or VUR should have early identification and individualized surgical treatment.
Proper bladder emptying should be employed early in treatment by CIC or urinary diversion with vesicostomy, preventing renal function deterioration. Individualized treatment warrants a better quality of life for these complex groups of patients, but the surgical team with versatility and skills in urological reconstruction is indispensable.
Level of Evidence: This is a case series (level IV of evidence)
Abbreviation list
CRS – Caudal Regression Syndrome
ARM – Anorectal malformation
VUR – Vesicoureteral reflux
VACTERL – Vertebral, anal, cardiac, tracheal, esophageal, renal, and limb anomalies.
MRI – Magnetic resonance images
CIC – Clean intermittent catheterization
MACE – Malone antegrade continence enema
US – Ultrasound
Declarations
Ethical approval: "The study was submitted and approved by the Research Ethics Committee of our Institution and the National Committee."
Informed Consent: "Informed consent was obtained from the guardians."
Funding sources: "The authors have no funding sources to declare."
Conflict of interests: "The authors have no conflicts of interest to declare."
Authorship: "All authors attest that they meet the current ICMJE criteria for Authorship."
References
- Duhamel B (1961) From the Mermaid to Anal Imperforation: The Syndrome of Caudal Regression. Arch Dis Child. 36(186): 152- 5.
- Rare disease database (2019) Caudal Regression Syndrome. NORD: National Organization for Rare Disorders.
- Welch JP, Aterman K (1984) The syndrome of caudal dysplasia: a review, including etiologic considerations and evidence of heterogeneity. Pediatr Pathol. 2(3): 313-27.
- Kylat RI, Bader M (2020) Caudal Regression Syndrome. Children (Basel). 7(11): 211.
- Seidahmed MZ, Abdelbasit OB, Alhussein KA, Miqdad AM, Khalil MI, et al. (2014) Sirenomelia and severe caudal regression syndrome. Saudi Med J. Suppl 1(Suppl 1): S36-43.
- Versiani BR, Gilbert-Barness E, Giuliani LR, Peres LC, Pina-Neto JM (2004) Caudal dysplasia sequence: severe phenotype presenting in offspring of patients with gestational and pregestational diabetes. Clin Dysmorphol. 13(1): 1-5.
- Kokrdova Z (2013) Caudal Regression Syndrome. J Obstet Gynaecol. 33(2): 202-3.
- Warner T, Scullen TA, Iwanaga J, Loukas M, Bui CJ, et al. (2020) Caudal Regression Syndrome-A Review Focusing on Genetic Associations. World Neurosurg. 138: 461-467.