


Aroon Singha1*, Syed Sameer Nasir MD2
1Collierville High School, Collierville, TN
2West Cancer Center, Memphis, TN
*Corresponding Author: Aroon Singha, Collierville High School, Collierville, TN.
Abstract
Carcinoid tumors originate from neuro-endocrine cells and can develop in organs such as the gastrointestinal tract, lungs, pancreas, and, rarely, in kidneys, breasts, ovaries, testicles, and other locations. The pathogenesis is uncertain as neuroendocrine cells are not found in renal parenchyma. We present a case of a 36-year-old female with a primary renal carcinoid tumor.
Keywords: primary renal carcinoid tumor; pleomorphic adenoma; 68Ga Dotatate PET scan; immunohistochemistry; SSTR2;
Case Report
A 36-year-old female patient was initially evaluated for painless hematuria and underwent CT abdomen and pelvis showing 1.9 cm mass in the medial aspect of her left lower kidney. The patient underwent a left partial nephrectomy, and the pathological evaluation revealed a well-differentiated neuroendocrine tumor (carcinoid tumor) measuring 2.3 cm [Figure 1]. Despite the generally low-grade nature of the tumor, it exhibited some high-risk features, including extension to the renal sinus fat area/perinephric fat and a small area of necrosis. One month after the partial nephrectomy, a follow-up CT scan revealed symmetric dense calcifications in the adrenal glands with no evidence of recurrence of disease. Thirteen months later, a CT scan showed a 2.1 cm mass adjacent to the left internal jugular vein and the left facial vein. Ga 68 dotatate positron emission tomography (PET) scan showed focal uptake in right parotid gland [Figure 2]. The patient then underwent a left parotidectomy, and the pathological examination revealed a pleomorphic adenoma of the parotid gland, comprising five tissue fragments up to 1.5 cm in size.
Discussion
[1] Primary renal carcinoid tumors are a rare form of neuroendocrine tumor. The tumors are present in adults with a mean age of diagnosis of 50, and predilection for either sex. Due to their rarity, the diagnosis and management of primary renal carcinoid tumors can be challenging, often requiring a multidisciplinary approach involving radiologists, pathologists, and urologic oncologists. We present a case of a 36-year-old female with a primary renal carcinoid tumor.
Radiological imaging, including computed tomography (CT) and magnetic resonance imaging (MRI), plays a crucial role in the initial evaluation and characterization of primary renal carcinoid tumors. [2] These modalities can help identify the location, size, and extent of the tumor, as well as any potential metastatic disease. Histopathological examination of the tumor tissue, obtained through biopsy or surgical resection, is essential for confirming the diagnosis and determining the appropriate treatment approach. [3] Immunohistochemical staining for neuroendocrine markers, such as chromogranin A and synaptophysin, is typically used to support the diagnosis.
Pathologically, primary renal carcinoid tumors are characterized by nests or sheets of monotonous, small to medium-sized cells with round to oval nuclei and granular cytoplasm. [4] These cells exhibit a distinctive neuroendocrine morphology and often stain positively for neuroendocrine markers, such as chromogranin A, synaptophysin, and CD56. The tumors may also exhibit various architectural patterns, including solid, trabecular, or glandular arrangements, and may show areas of necrosis or hemorrhage.
The histological grading of primary renal carcinoid tumors is based on the mitotic rate, presence of necrosis, and the Ki-67 proliferation index, like other neuroendocrine tumors. [6] Well-differentiated tumors typically have a low mitotic rate and Ki-67 index, while poorly differentiated tumors exhibit higher proliferative activity and more aggressive behavior. Accurate grading is crucial for determining the appropriate treatment approach and predicting prognosis.
[5] The treatment of primary renal carcinoid tumors is challenging due to their rarity. Surgery is the primary treatment for localized tumors, aiming to remove the tumor while preserving kidney function. For advanced or metastatic cases, additional therapies like somatostatin analogs, targeted therapies, or chemotherapy may be considered, although their effectiveness is not well-established. Close monitoring and developing new targeted therapies are crucial for improving outcomes in these rare tumors. In our case, the patient underwent a partial nephrectomy with negative margins, and we decided to observe.
[1] The outlook for primary renal carcinoid tumors varies significantly and depends on factors like the stage at diagnosis, tumor grade, and whether the cancer has spread. Generally, localized and low-grade tumors have a better prognosis when completely removed through surgery. Our patient has had a surveillance scan annually, and there has been no evidence of recurrence of the disease.
[7] Another interesting finding in our case is the incidental and strong focal tracer uptake in a pleomorphic adenoma (PA) in right parotid gland undergoing left parotidectomy confirming false positive 68 Ga- Dotatate PET scan result. [8] This has been described previously and is related to the strong presence of the somatostatin receptor 2 (SSTR2) in PA on immunohistochemistry. [8] An analysis of 306 tumors including 207 non-PA and 99 PA using the HER2-mama scale for the assessment of SSTR2 expression demonstrated that PA strongly expresses the SSTR2. This provides a biomolecular explanation for the tracer uptake and false positive result in the PA described in our case.
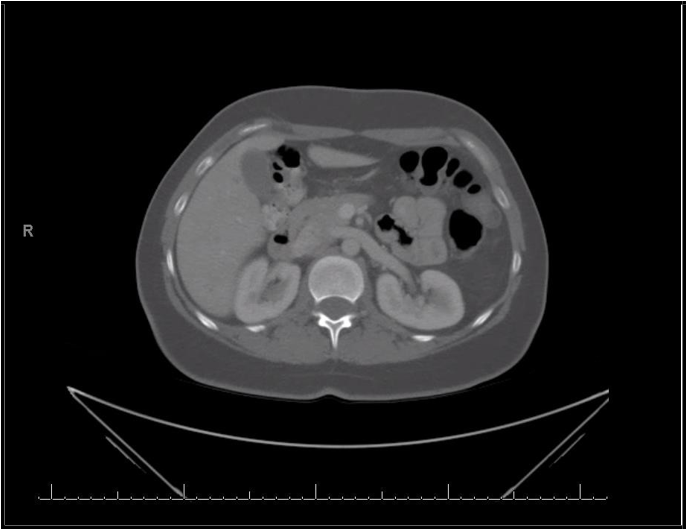
Figure 1: Left kidney carcinoid tumor
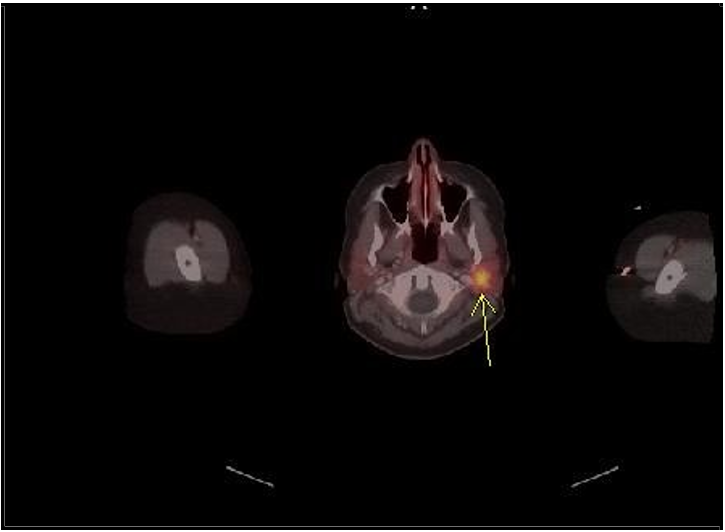
Figure 2: 68 Ga Dotatate PET scan showing focal tracer uptake in the left parotid gland
Acknowledgements: Pathologist to provide readings.
Author Contributions: AS: writing of original draft, review and editing. SN: provided data, review and editing.
References
- Kanthan R, Senger JL, Kanthan S (2017) Primary carcinoid tumor of the kidney: A report of two cases and review of literature. World J Surg Oncol. 15(1): 15.
- Armah HB, Parwani AV, Perepletchikov AM (2011) Primary carcinoid tumor of the kidney: A case report and review of the literature. Diagn Pathol. 6: 15.
- Jeung JA, Cao D, Selli C, Hinds S, Lam AP. Primary carcinoid tumor of the kidney: A case report and review of the literature. Curr Urol. 2012;6(3): 162-164.
- Omiyale AX, Venyo AK. Primary carcinoid tumor of the kidney: A review of the literature. Adv Urol. 2015;2015: 179812.
- Yoo J, Park S, Jung Lee H, Jin Kang S, Kee Kim B (2010) Primary carcinoid tumor arising in a horseshoe kidney: A case report and review of the literature. Pathol Int. 60(7): 537-544.
- Jhala N, Jhala D, Eloubeidi MA, et al. (2003) Renal carcinoid: a case report and literature review. Am J Clin Pathol. 119(1): 135- 141.
- Laurens ST, Netea-Maier RT, Aarntzen EJHG (2018) 68Ga- DOTA-TOC Uptake in Pleomorphic Adenoma. Clinical Nuclear Medicine 43(7): 524-525.
- Johnson F, Hofauer B, Wirth M, Wollenberg B, Stögbauer F, et al. (2023) Novel Discovery of the Somatostatin Receptor (SSTR2) in Pleomorphic Adenomas via Immunohistochemical Analysis of Tumors of the Salivary Glands. Cancers. 15(15): 3917.