


Brian Behnke1*, Kavanya Feustel1, Scott Mayer1, Nikhilesh Srinivasan1, Dmitri Scherbak2
1Internal Medicine Resident at HCA Health One- Sky Ridge Medical Center
2Internal Medicine Research Director at HCA Health One- Sky Ridge Medical Center
*Corresponding Author: Brian Behnke, Internal Medicine Resident at HCA Health One- Sky Ridge Medical Center
Introduction
Hypocalcemia can present with a wide variety of nonspecific symptoms including seizures, paresthesia’s, encephalopathy, psychosis, dry skin, dysphagia and prolonged QT interval. [1,2] There are many acute and chronic etiologies, including renal failure, parathyroid hormone (PTH) resistance, hypoparathyroidism, vitamin D deficiency, disorders of magnesium metabolism and acute pancreatitis. Pseudohypoparathyroidism (PHP) is a rare congenital disorder with an estimated incidence of 1 per 100,000 people in the United States.[3,4] PHP pathophysiology is due to target organ resistance, especially renal resistance to PTH, impairing the formation of active Vitamin D and reducing sodium-dependent phosphate transporters leading to elevated PTH, hypocalcemia, and hypophosphatemia.[3] There are multiple subtypes of PHP. Types 1a and 1c exhibit PTH, TSH and Gn resistance, 1a with associated GHRH resistance. Both type 1 PHP patients have features of Albright hereditary osteodystrophy, characterized by short, stocky stature, early-onset obesity, brachydactyly of the metatarsals, broad distal phalanges or acrodysostosis, ectopic ossifications and neurodevelopmental deficits.[3,5].
This case study aims to highlight PHP including clinical presentation, symptoms and complications of medical management of the rare disease. By learning more about PHP, we can gain a better understanding of how to medically manage those patients it affects.
Case Report
A 60 year-old male with a past medical history of PHP type 1a, peripheral artery disease status-post stenting, hypothyroidism and mild cognitive delay presented to the emergency department via family after two transient, unresponsive staring episodes and 24 hours of increasing confusion. He exhibited absence-like seizures with post-ictal state of a baseline mild encephalopathy, no longer being able to name his medications, drive, or carry a conversation. The patient endorsed poor medication compliance over the prior weeks, supported by an inappropriately full dosette box.
On presentation he was mildly hypertensive to 179/100. Initial labs revealed neutrophilic predominant leukocytosis 11.4 10^9/L, hypocalcemia of 7 mg/dL, normal albumin of 4, elevated alkaline phosphatase 279 U/L. Follow-up ionized calcium was 0.90 mmol/L [Normal 1.12-1.32]. Magnetic resonance imaging (MRI) of the brain, electroencephalogram (EEG), computed tomography angiography (CTA) of the head and neck were all negative for acute abnormalities. He was treated with intravenous (IV) and oral calcium supplementation along with calcitriol and ergocalciferol. From which he returned to baseline by discharge 1 week later.
When probed, the patient endorsed intentional lapses in calcium supplementation due to its previous adverse effects of a myocardial infarction requiring stenting and other peripheral vessel occlusions as he wished to avoid these painful sequelae.
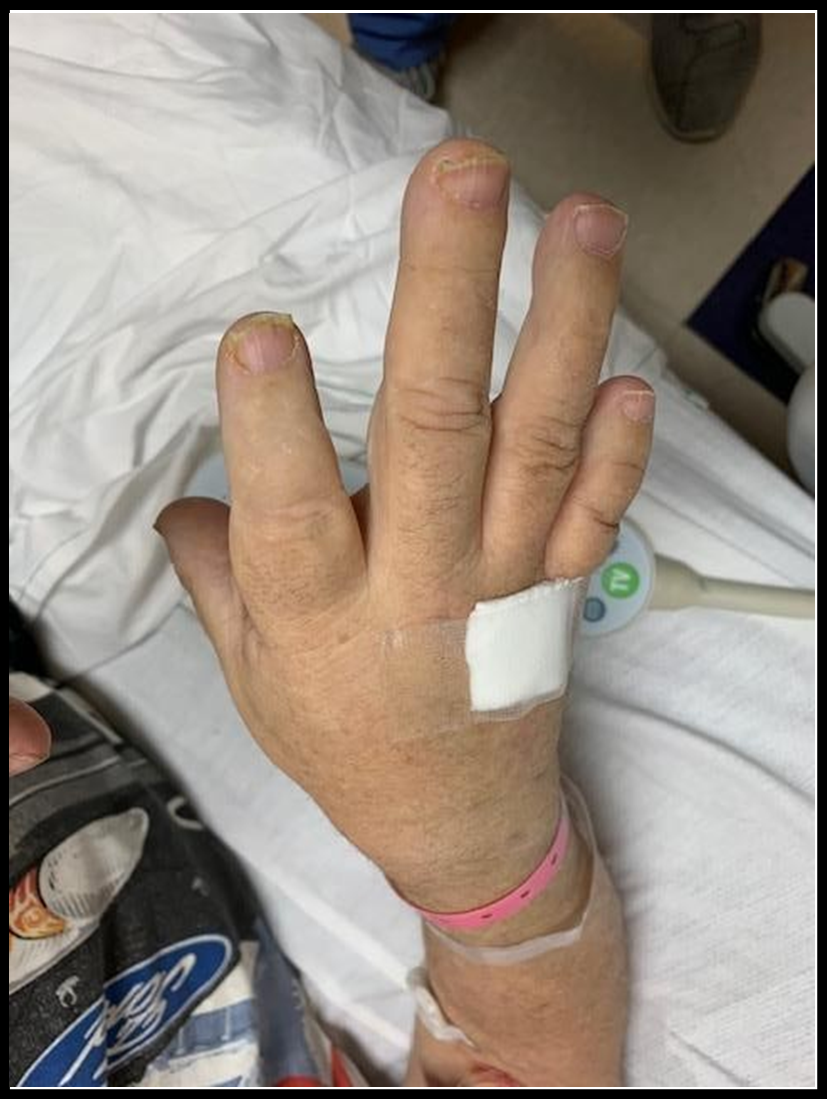
Figure 1: Brachydactyly of Right Hand (Dorsal)
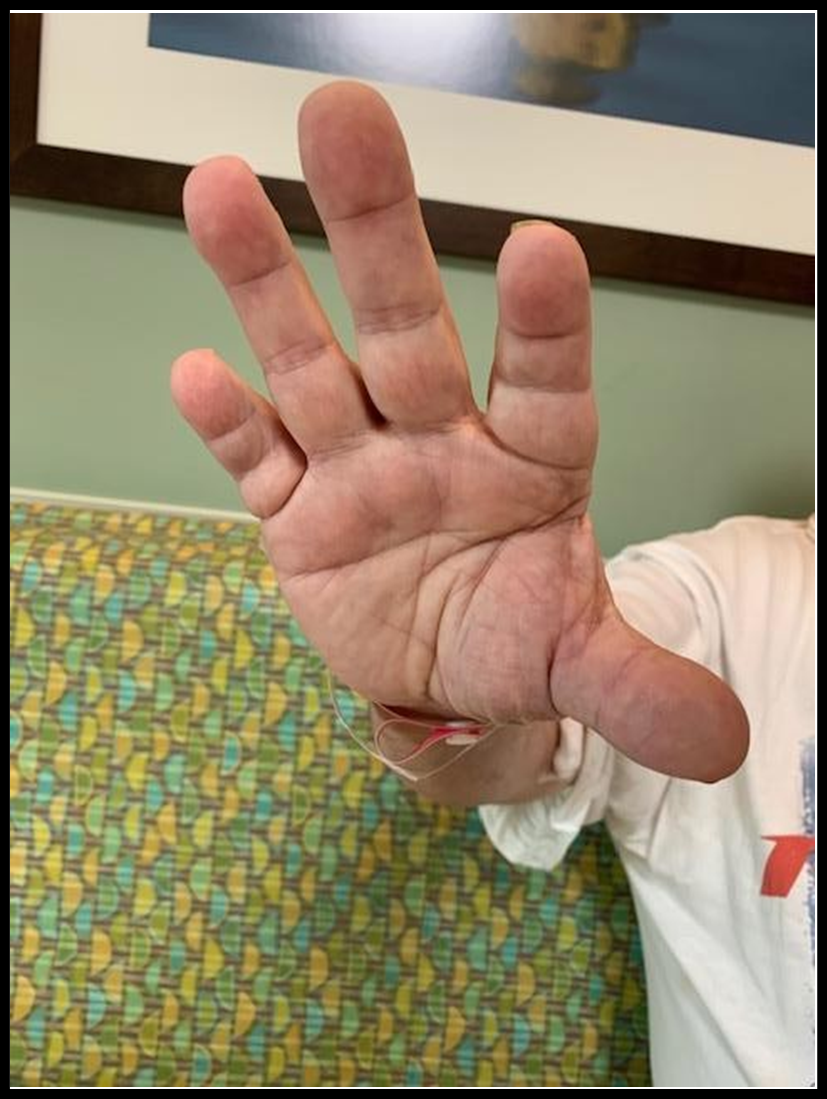
Figure 2: Brachydactyly of Right Hand (Palmar)
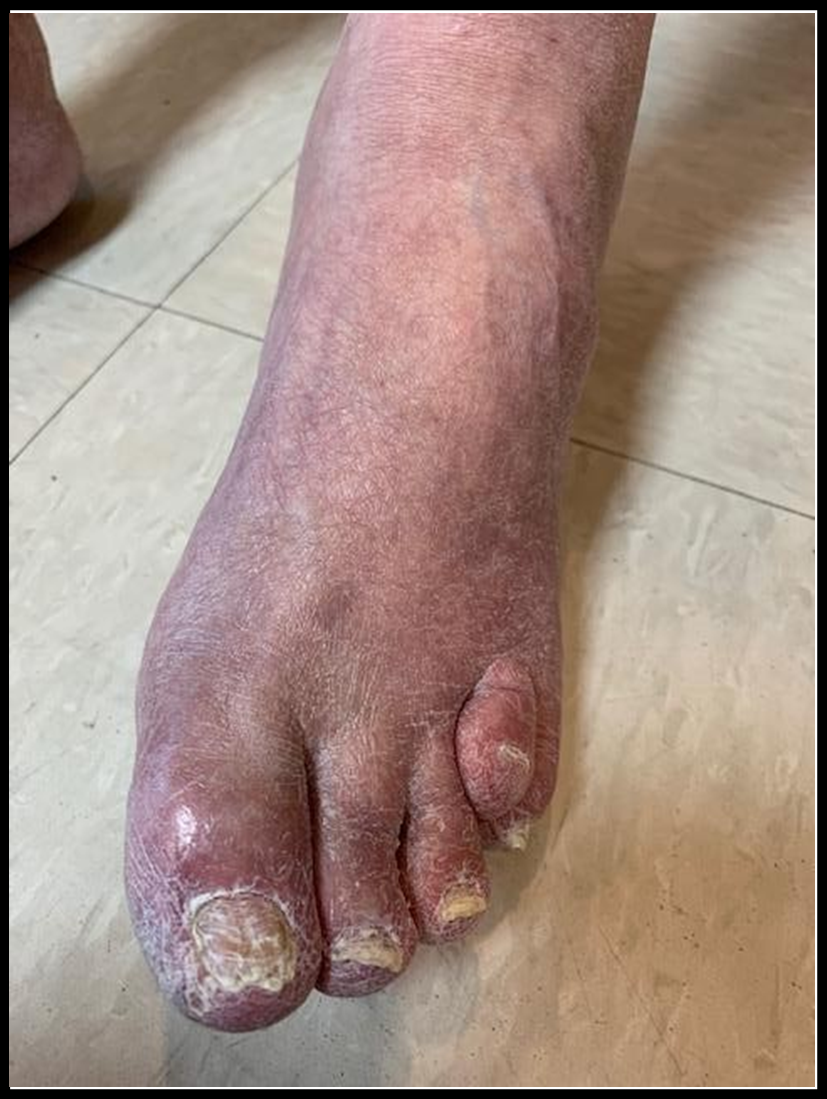
Figure 3: Acrodysostosis of Left Foot
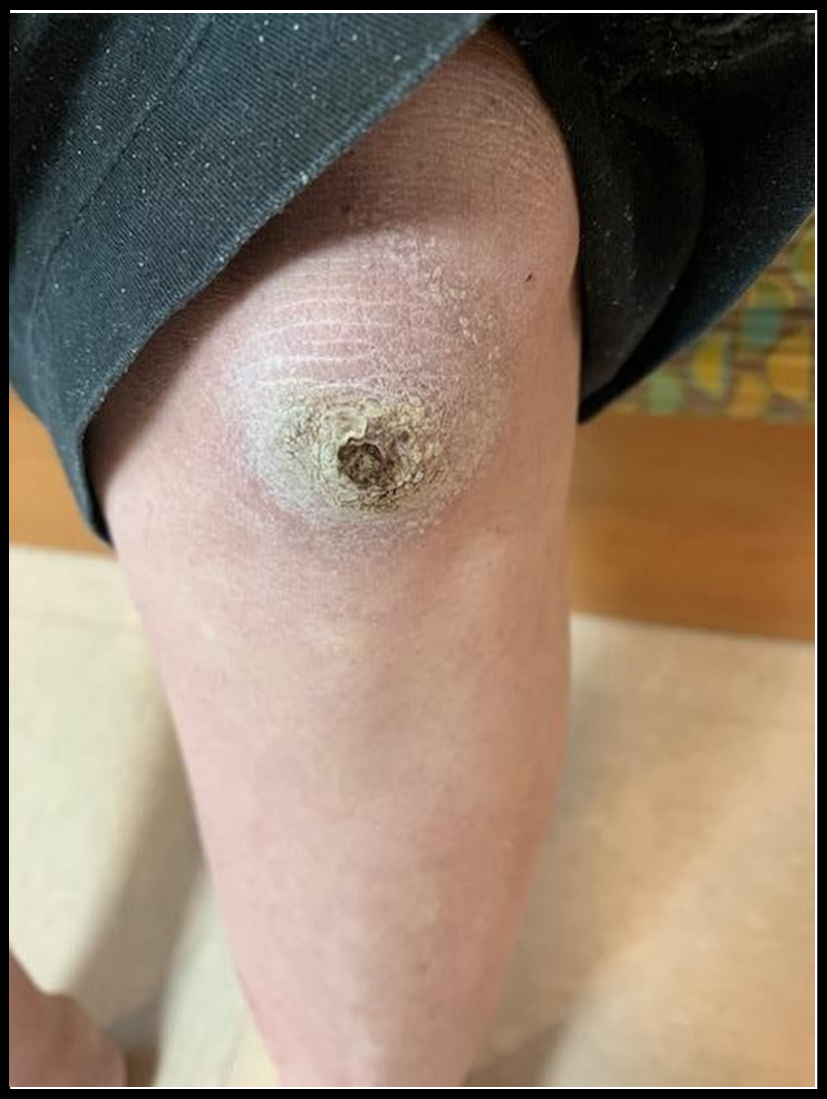
Figure 4: Subcutaneous ossification of Left Knee
Discussion
Hypocalcemia with elevated PTH can be attributed to malabsorption, organ dysfunction, deficient intake, or various hereditary resistances. In this case, due to an unknown genetic abnormality but phenotypic type 1 PHP, the patient most likely has 1a PHP associated with multihormonal target tissue resistance, attributed to loss of function of maternal guanine nucleotide-binding protein gene (GNAS).[5,6] Chronic hypocalcemia in patients with PHP can cause extreme consequences such as in our patient with seizures, a not-uncommon adverse effect.5 Case reports outline a female with hypocalcemia, prolonged QT interval, and generalized tonic-clonic seizures and a 64-year-old female with PHP1a who was found to have basal ganglia calcifications, leading to generalized seizures.[4,7]
Management of this disease can be very challenging. It requires routine monitoring of serum PTH, calcium, phosphate, and calcifediol with more frequent monitoring in patients who are symptomatic. Severe, symptomatic hypocalcemia requires immediate intervention with IV calcium, as seen with our patient. Treatment with active vitamin D metabolites or analogs is recommended. It is important to perform regular renal imaging to evaluate for nephrocalcinosis as well. Phosphate binders are adequate to treat associated hyperphosphatemia.[8] This lifelong disease requires a complex multifactorial treatment and monitoring approach to offer the best quality of life to those it affects.
Conclusion
PHP is a rare disease typically diagnosed in childhood. Mainstays of treatment include calcitriol and calcium supplementation, which can lead to painful adverse effects due to peripheral accumulation like nephrolithiasis and vascular diseases as seen in this patient. Balancing patient wellbeing from hypocalcemia and the significant calcium supplementation required is a conundrum that has yet to be solved. Shared decision-making and consideration of the patient’s comfort are paramount to achieving therapeutic success.
Informed Consent: Consent was obtained from the patient.
References
- Schafer AL, Shoback DM (2000) Hypocalcemia: Diagnosis and Treatment. [Updated 2016 Jan 3]. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet].
- Mantovani G, Bastepe M, Monk D, de Sanctis L, Thiele S, et al. (2020) Recommendations for Diagnosis and Treatment of Pseudohypoparathyroidism and Related Disorders: An Updated Practical Tool for Physicians and Patients. Horm Res Paediatr. 93(3): 182-196.
- Clarke BL, Brown EM, Collins MT, Jüppner H, Lakatos P, et al. (2016) Epidemiology and Diagnosis of Hypoparathyroidism. J Clin Endocrinol Metab. 101(6): 2284-2299.
- Del Monte P, Cuttica CM, Marugo A, Foppiani L, Audenino D, et al. (2019) Unrecognized Pseudohypoparathyroidism Type 1A as a Cause of Hypocalcemia and Seizures in a 64-Year-Old Woman. Case Rep Endocrinol. 2019: 8456239.
- Najim MS, Ali R, Awad M, Omer A (2020) Pseudohypoparathyroidism presenting with seizures: a case report and literature review. Intractable Rare Dis Res. 9(3): 166-170.
- Mantovani G, Bastepe M, Monk D, de Sanctis L, Thiele S, et al. (2018) Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nat Rev Endocrinol. 14(8): 476-500.
- Hendy GN, Cole DEC, Bastepe M (2000) Hypoparathyroidism and Pseudohypoparathyroidism. [Updated 2017 Feb 19]. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.
- Shoemaker AH, Jüppner H (2017) Nonclassic features of pseudohypoparathyroidism type 1A. Curr Opin Endocrinol Diabetes Obes. 24(1): 33-38.