


Oğuzhan Yıldız1, Seda Yılmaz2*, Özcan Çeneli3, Sinan Demircioğlu3, Atakan Tekinalp3
1 Necmettin Erbakan University, Meram Faculty of Medicine, Department of Oncology, Konya, Turkey
2 Konya City Hospital, Department of Internal Medicine, Clinic of Hematology, Konya, Turkey
3 Necmettin Erbakan University, Meram Faculty of Medicine, Department of Hematology, Konya, Turkey
*Corresponding Author: Seda Yılmaz, Konya City Hospital, Department of Internal Medicine, Clinic of Hematology, Konya, Turkey.
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory disease. Infectious causes can be triggers. While it is mainly seen after viral infections, it can be seen after bacteria, parasite, and fungal infections to a lesser extent. This article presented a case of HLH triggered by pulmonary aspergillosis.
Keywords: Aspergillosis, hemophagocytic lymphohistiocytosis, pancytopenia
Introduction
Hemophagocytic lymphohistiocytosis is an aggressive and life-threatening syndrome that can occur at any age due to the overactivation of the immune system. It can be seen as familial or sporadic. It can be triggered by various events that disrupt immune homeostasis.
Infection can be a trigger both in cases with genetic tendencies and in sporadic cases. It is essential to initiate the treatment early. Since it is a rare syndrome, variable clinic presentation and non-specific laboratory findings may cause delays in diagnosis.
Case Report
A 28-year-old female patient applied at the emergency unit complaining of weakness, fatigue, blackout, fever, and cough continuing for 3 weeks. The patient was diagnosed with pancytopenia and was admitted to our hematology department for further investigation of fever and pancytopenia. In the complete blood count, leukocyte was 1.800/μL, neutrophil 1.200/μL, hemoglobin 7.3 g/dL, platelet 54.0×103/μL; in the biochemical tests, and renal function tests were regular, AST:119 (5-34) U/L, ALT: 44 (0-55) U/L, triglyceride 370 mg/dL, fibrinogen: 144 mg/dL, ferritin: 1650 ng/mL. Hepatitis A virus-specific IgM antibody, hepatitis B surface antigen, hepatitis C virus antibody, viral capsid antigen IgM, cytomegalovirus antigen, and HIV antibody were negative. Rheumatologic examination and immunological markers were negative. Liver enzymes increased to 690 U/L during follow-up. Aspergillus galactomannan antigen level (ELISA) was found to be 1.76. On abdominal ultrasonography (USG), the liver was 162 mm, and the spleen was 155 mm. Superficial tissue USG showed no lymph nodes. Pulmonary tomography showed patchy peribronchial ground glass density and diffuse nodules in the right and left lung parenchyma (Figure 1).
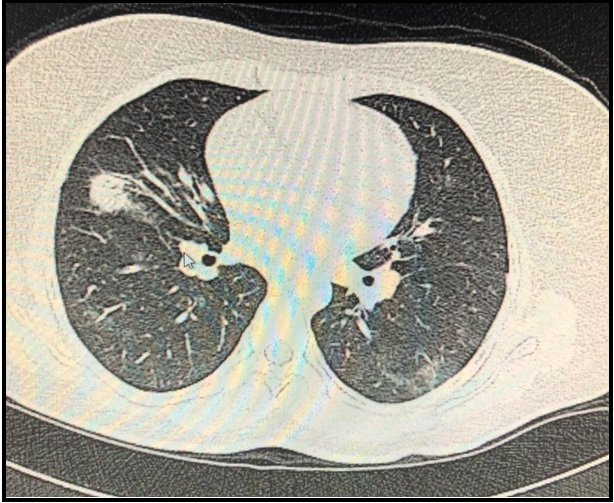
Figure-1: Torax CT images
Bone marrow biopsy showed no microorganism in PAS staining, and although CD68 + multiple histiocytes were present, no clear hemophagocytosis was observed. The hemophagocytic syndrome was considered as five of the eight clinical and laboratory diagnostic criteria accepted by the hemophagocytic syndrome study group were available. HLH-2004 protocol and voriconazole treatment were initiated. After 12 weeks, nodules disappeared in the control thoracic computerized tomography. In addition, cytopenia improved, and the liver and spleen regressed to standard size. The patient with improved clinical and laboratory findings is being followed up in remission.
Discussion
Hemophagocytic lymphohistiocytosis (HLH) is a fatal clinical syndrome within the histiocytosis group. HLH is divided into two groups, genetic and acquired; acquired HLH can occur in all age groups. Viral, bacterial, fungal, and parasitic diseases, malignancy, metabolic diseases, immunodeficiency and collagen tissue disease, nonsteroidal anti-inflammatory drugs, methotrexate, and gold salts may be responsible for the etiology of HLH [1-3]. Nowadays, cases of HLH associated with infection are increasingly reported. It is generally associated with viral infections such as EBV, CMV, Parvovirus, HSV, VZV, measles, HHV-8, H1N1 influenza virus, and HIV, and may be seen to a lesser extent after bacterial, parasitic, and fungal infections [4-13]. Several cases of fungal infection have been reported in the literature [14]. In our case, invasive Aspergillus was one of the rare causes.
Conclusion
As a result, in treating a patient with HLH, the critical step is to suppress severe inflammation, which is responsible for life- threatening symptoms. If a curable organism can be identified, treatment with this pathogen can cause a rapid remission of the possibly fatal hemophagocytosis.
Disclaimer: None.
Conflict of Interest: None.
Funding Sources: None.
References
- Hadchouel M, Prieur AM, Griscelli C (1985) Acute hemorrhagic,hepatic and neurologic in juvenile idiopathic arthritis :possible reaction to drugs and infections. J Pediatr. 106(4): 561-6.
- Leal FE, Cavazzana CL, de Andrade HF Jr, Galisteo A Jr, de Mendonça JS, et al. (2007) Toxoplasma gondii pneumonia in immunocompetent subjects: case report and review. Clin Infect Dis. 44(6): e62-6.
- Rigante D, Capoluongo E, Bertoni B, Ansuini V, Chiaretti A, et al. (2007) First report of macrophage activation syndrome in hyperimmunoglobulinemia-D with periodic fever syndrome. Arthritis Rheum. 56(2): 658-61.
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X (2014) Adult haemophagocytic syndrome. Lancet. 383(9927): 1503-1516.
- McClain K, Gehrz R, Grierson H, Purtilo D, Filipovich A (1988) Virus-associated histiocytic proliferations in children. Frequent association with Epstein-Barr virus and congenital or acquired immunodeficiencies. Am J Pediatr Hematol Oncol. 10(3): 196- 205.
- Mou SS, Nakagawa TA, Riemer EC, McLean TW, Hines MH, et al. (2006) Hemophagocytic lymphohistiocytosis complicating influenza A infection. Pediatrics. 118(1): e216-9.
- Harms PW, Schmidt LA, Smith LB, Newton DW, Pletneva MA, et al. (2010) Autopsy findings in eight patients with fatal H1N1 influenza. Am J Clin Pathol. 134(1): 27-35.
- Yuzurihara SS, Ao K, Hara T, Tanaka F, Mori M, et al. (2013) Human parechovirus-3 infection in nine neonates and infants presenting symptoms of hemophagocytic lymphohistiocytosis. J Infect Chemother. 19: 144–148.
- Chen TL, Wong WW, Chiou TJ (2003) Hemophagocytic syndrome: an unusual manifestation of acute human immunodeficiency virus infection. Int J Hematol. 78(5): 450-2.
- Fardet L, Blum L, Kerob D, Agbalika F, Galicier L, et al. (2003) Human herpesvirus 8-associated hemophagocyticlymphohistiocytosis in human immunodeficiency virus-infected patients. Clin Infect Dis. 37(2): 285-91.
- Grossman WJ, Radhi M, Schauer D, Gerday E, Grose C, et al. (2005) Development of hemophagocytic lymphohistiocytosis in triplets infected with HHV-8. Blood. 106(4): 1203–1206.
- Hegerova LT, Lin Y (2013) Disseminated histoplasmosis: a cause of hemophagocytic syndrome. Mayo Clin Proc. 88(10): e123.
- Otrock ZK, Eby CS (2015) Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. 90(3): 220-4.
- Majima S, Okachi S, Asano M, Wakahara K, Hashimoto N, et al. (2018) “Pseudomembranous Invasive Tracheobronchial Aspergillosis with Fulminant Hepatitis and Hemophagocytic Syndrome." Internal Medicine. 57(16): 2371–2375.